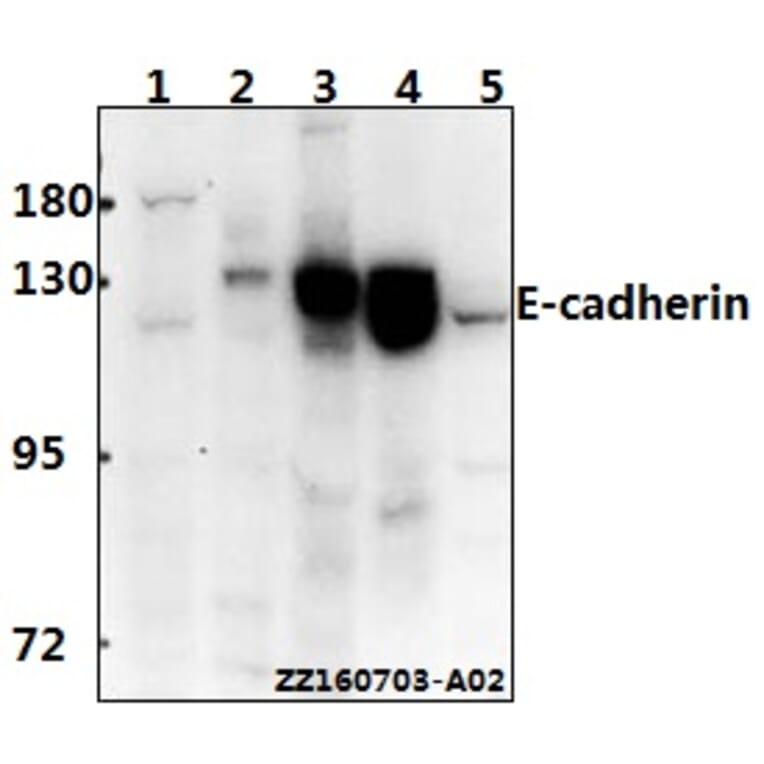
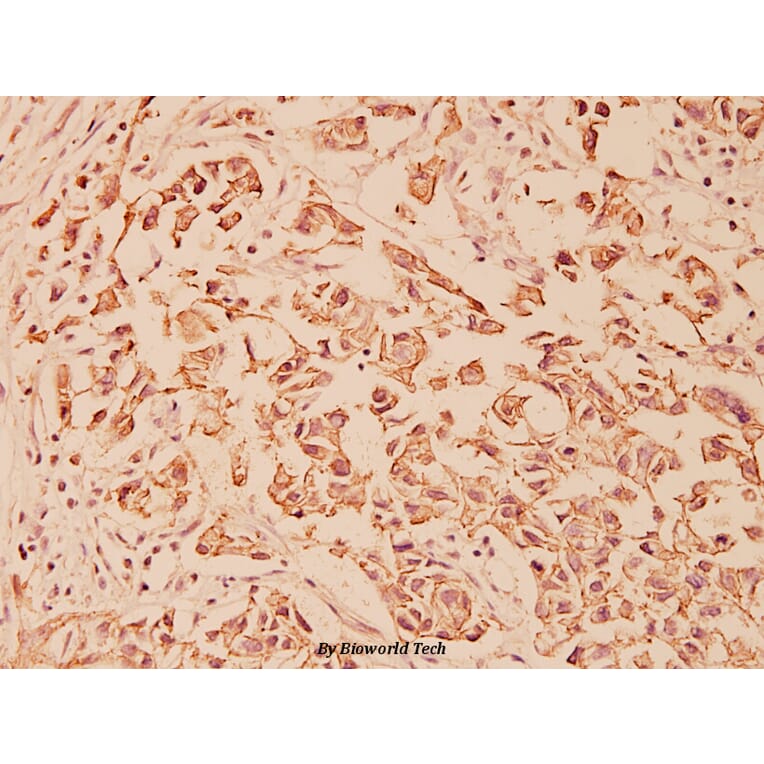
Unconjugated
Research on the relationship between aberrant long non-coding RNA (lncRNA) and cancer stem cell (CSC) biology in cancer patients has been recently gaining attention. The goal of this study was to investigate whether the decreasing lncRNA HOTAIR expression would inhibit human colorectal cancer (CRC) stem cells. CD133(+)CSCs were isolated from human CRC LoVo cell line by using a magnetic-activated cell sorting system, and were transfected with the expression vector-based small hairpin RNA targeting HOTAIR (shHOTAIR). The ability of cellular proliferation, migration, invasion, colony-forming, and the epithelial-mesenchymal transition (EMT)-associated molecule expression as well as the tumorigenicity of CD133(+)-shHOTAIR were evaluated by the MTT, wound-healing, cellular invasion, colony formation and Western blot assays, respectively. This study found that, when compared with control cells in vitro, CD133(+)-shHOTAIR exhibited the decreased HOTAIR expression, suppressed cellular proliferation, migration, invasion, colony-forming, and inhibited the Vimentin expression with increased E-cadherin expression. In particular, the down-regulation of the HOTAIR expression in CD133(+)CSCs markedly attenuated the tumor growth and lung metastasis in xenograft nude mice. Taken together, this study found that down-regulating the HOTAIR expression in CD133(+)CSCs could serve as a potential anti-cancer regimen to inhibit the invasiveness and metastasis of CRC CSCs.
Tumor vaccines may induce antitumor efficacy, however, weak immunogenicity of tumor antigens is one of the prime obstacles for excitation of the antitumor immune responses. Therefore, strategies that enhance immunogenicity of tumor vaccines are of particular interest. In this study, a novel melanoma B16F10 CD133(+)CD44(+) cancer stem cell (CSC) vaccine expressing 6 kDa early secreted antigenic target (ESAT-6) in the glycosylphosphatidylinositol (GPI)-anchored form and secreting interleukin (IL)-21 was developed. Its anti-melanoma efficacy and mechanisms were investigated in mice. The results demonstrated that the B16F10-ESAT-6-gpi/IL-21 CD133(+)CD44(+) CSC vaccine exhibited enhanced anti-melanoma efficacy as determined by inhibited melanoma growth, prolonged survival of melanoma bearing mice. The anti-melanoma immunity was associated with elevated levels of serum anti-ESAT-6 and interferon (IFN)-γ as well as increased cytotoxic activities of natural killer cells, splenocytes, and complement dependent cytotoxicity. Furthermore, this CSC-based vaccine apparently inhibited melanoma lung metastasis by decreasing the level of Vimentin while increasing the level of E-cadherin expression, suggesting an inhibited epithelial mesenchymal transition. Thus, the B16F10-ESAT-6-gpi/IL-21 CD133(+)CD44(+) CSC vaccine may be used to reactivate the anti-tumor immunity and for treatment of melanoma.
Urinary bladder cancer (UBC) patients at muscle invasive stage have poor clinical outcome, due to high propensity for metastasis. Cancer-associated fibroblasts (CAFs), one of the principal constituents of the tumor stroma, play an important role in tumor development. However, it is unclear whether CAFs from UBC induce cell invasion and which signaling pathway is involved. Herein, we found that conditional medium from UBC CAFs (CAF-CM) enhanced the invasion of UBC cells. CAF-CM induced the epithelial-mesenchymal transition (EMT) by regulating expression levels of EMT-associated markers in UBC cells. Higher concentration of TGFβ1 in CAF-CM, comparing with the CM from adjacent normal fibroblast, led to phosphorylation of Smad2 in UBC cells. Additionally, inhibition of TGFβ1 signaling decreased the EMT-associated gene expression, and cancer cell invasion. Interestingly, a long non-coding RNA, ZEB2NAT, was demonstrated to be essential for this TGFβ1-dependent process. ZEB2NAT depletion reversed CAF-CM-induced EMT and invasion of cancer cells, as well as reduced the ZEB2 protein level. Consistently, TGFβ1 mRNA expression is positively correlated with ZEB2NAT transcript and ZEB2 protein levels in human bladder cancer specimens. Our data revealed a novel mechanism that CAFs induces EMT and invasion of human UBC cells through the TGFβ1-ZEB2NAT-ZEB2 axis.
The prognosis of patients with ovarian cancer has remained poor mainly because of aggressive cancer progression. Since epithelial-mesenchymal transition (EMT) is an important mechanism mediating invasion and metastasis of cancer cells, targeting the EMT process with more efficacious and less toxic compounds to inhibit metastasis is of great therapeutic value for the treatment of ovarian cancer. We have found for the first time that the ginsenoside 20(S)-Rg3, a pharmacologically active component of the traditional Chinese herb Panax ginseng, potently blocks hypoxia-induced EMT of ovarian cancer cells in vitro and in vivo. Mechanistic studies confirm the mode of action of 20(S)-Rg3, which reduces the expression of hypoxia-inducible factor 1α (HIF-1α) by activating the ubiquitin-proteasome pathway to promote HIF-1α degradation. A decrease in HIF-1α in turn leads to up-regulation, via transcriptional suppression of Snail, of the epithelial cell-specific marker E-cadherin and down-regulation of the mesenchymal cell-specific marker vimentin under hypoxic conditions. Importantly, 20(S)-Rg3 effectively inhibits EMT in nude mouse xenograft models of ovarian cancer, promising a novel therapeutic agent for anticancer therapy.
To support the high rates of proliferation, cancer cells undergo the metabolic reprogramming: aerobic glycolysis and glutamine addiction. Though glucose regulated protein 78 (GRP78) is a glucose-sensing protein and frequently highly expressed in tumor cells, its roles in glucose and glutamine metabolic regulation remain poorly unknown. We report here that glucose deficiency-induced GRP78 enhances β-catenin signaling and consequently promotes its downstream c-Myc-mediated glutamine metabolism in colorectal cancer cells. Mechanistically, GRP78 elevates intracellular free β-catenin level via disruption of adenomatous polyposis coli (APC)-β-catenin and E-cadherin-β-catenin protein complexes. Notably, overexpression of GRP78 causes APC protein downregulation in proteasome- and lysosome-independent manners. Further mechanistic studies reveal that GRP78 facilitates the extracellular release of APC, thereby rendering the liberation of β-catenin from APC. Furthermore, GRP78 acts through both hindering E-cadherin expression and impairing the interaction of E-cadherin with β-catenin to indirectly and directly influence E-cadherin-β-catenin complex stability. Our study reveals that GRP78 is a novel molecular link between metabolic alterations and signal transduction during tumor progression.
Gastric cancer (GC) is the fourth most common malignancy in males and the fifth most common malignancy in females worldwide. DACH1 is frequently methylated in hepatic and colorectal cancer. To further understand the regulation and mechanism of DACH1 in GC, eight GC cell lines, eight cases of normal gastric mucosa, 98 cases of primary GC and 50 cases of adjacent non-tumour tissues were examined. Methylation-specific PCR, western blot, transwell assay and xenograft mice were used in this study. Loss of DACH1 expression correlated with promoter region methylation in GC cells, and re-expression was induced by 5-Aza-2'-deoxyazacytidine. DACH1 is methylated in 63.3% (62/98) of primary GC and 38% (19/50) of adjacent non-tumour tissues, while no methylation was found in normal gastric mucosa. Methylation of DACH1 correlated with reduced expression of DACH1 (P < 0.01), late tumour stage (stage III/IV) (P < 0.01) and lymph node metastasis (P < 0.05). DACH1 expression inhibited epithelial-mesenchymal transition and metastasis by inhibiting transforming growth factor (TGF)-β signalling and suppressed GC cell proliferation through inducing G2/M phase arrest. The tumour size is smaller in DACH1-expressed BGC823 cell xenograft mice than in unexpressed group (P < 0.01). Restoration of DACH1 expression also sensitized GC cells to docetaxel. These studies suggest that DACH1 is frequently methylated in human GC and expression of DACH1 was controlled by promoter region methylation. DACH1 suppresses GC proliferation, invasion and metastasis by inhibiting TGF-β signalling pathways both in vitro and in vivo. Epigenetic silencing DACH1 may induce GC cells' resistance to docetaxel.
Liver metastasis is a major cause of mortality in colorectal cancer (CRC). The current study was to investigate the ability of ulinastatin (UTI) and curcumin (CUR) to inhibit CRC liver metastases via modulating matrix metalloproteinase-9 (MMP-9) and E-cadherin expression. Human CRC HCT-116 cells were treated with compounds individually and in combination in order to understand the effect on cell migration and invasion. The HCT-116 cell line was established to stably express luciferase and green fluorescent protein (GFP) by lentiviral transduction (HCT-116-Luc-GFP). We identified an anti-metastasis effect of UTI and CUR on a CRC liver metastasis mouse model. Tumor development and therapeutic responses were dynamically tracked by bioluminescence imaging. Expression of MMP-9 and E-cadherin in metastatic tumors was detected by immunohistochemical assay. Results of wound healing and cell invasion assays suggest that treatment with UTI, CUR, and UTI plus CUR, respectively, significantly inhibit HCT-116 cell migration and invasion. Furthermore, results of CRC hepatic metastasis on a nude mouse model showed that treatment with UTI, CUR alone, and a combination notably inhibited hepatic metastases from CRC and prolonged survival of tumor-bearing mice, especially in the UTI plus CUR group. These results suggest that the combination of UTI and CUR together may offer greater inhibition against metastasis of CRC.
We investigated the protective effect of benidipine, by testing the changes of the activity of Rho kinase and transdifferentiation of renal tubular epithelium cells in vivo. Wistar rats were randomly divided into two groups: normal (N) and diabetes. STZ were used to make the rats type 1 diabetic and were randomly assigned as diabetes without treatment (D), diabetes treated with benidipine (B), and diabetes treated with fasudil (F) and treated for 3 months. Immunohistochemistry and western blotting were for protein expressions of ROCK1, α-SMA, and E-cadherin and real-time PCR for the mRNA quantification of ROCK1. Compared with N group, D group had significant proliferation of glomerular mesangial matrix, increased cell number, thickened basement membrane, widely infiltrated by inflammatory cells and fibrosis in the renal interstitial, and dilated tubular. Those presentations in F and B groups were milder. Compared with N group, D group showed elevated MYPT1 phosphorylation, increased expression of ROCK1, α-SMA protein, and ROCK1 mRNA and decreased expression of E-cadherin protein. B group showed attenuated MYPT1 phosphorylation, decreased ROCK1, α-SMA protein, and ROCK1 mRNA expression and increased expression of E-cadherin protein. In conclusion, benidipine reduces the epithelium-mesenchymal transdifferentiation and renal interstitial fibrosis in diabetic kidney by inhibiting ROCK1 activity.
Pulmonary fibrosis is a progressive and fatal fibrotic disease of the lungs with unclear etiology. Recent insight has suggested that early injury/inflammation of alveolar epithelial cells could lead to dysregulation of tissue repair driven by multiple cytokines. Although dysregulation of interleukin- (IL-) 22 is involved in various pulmonary pathophysiological processes, the role of IL-22 in fibrotic lung diseases is still unclear and needs to be further addressed. Here we investigated the effect of IL-22 on alveolar epithelial cells in the bleomycin- (BLM-) induced pulmonary fibrosis. BLM-treated mice showed significantly decreased level of IL-22 in the lung. IL-22 produced γδ T cells were also decreased significantly both in the tissues of lungs and spleens. Administration of recombinant human IL-22 to alveolar epithelial cell line A549 cells ameliorated epithelial to mesenchymal transition (EMT) and partially reversed the impaired cell viability induced by BLM. Furthermore, blockage of IL-22 deteriorated pulmonary fibrosis, with elevated EMT marker ( α -smooth muscle actin ( α -SMA)) and overactivated Smad2. Our results indicate that IL-22 may play a protective role in the development of BLM-induced pulmonary fibrosis and may suggest IL-22 as a novel immunotherapy tool in treating pulmonary fibrosis.
Epithelial-mesenchymal transition (EMT) plays a specific role in the migration of tumor cells. Both estrogen and midkine (MK) have been thought to be important factors in promoting the progression of non-small-cell lung cancer (NSCLC) and can enhance EMT. Some evidence indicated the correlation between estradiol (E2) and MK, but the precise mechanism on their interreaction is unknown. Here, we try to clarify whether and how E2 regulates MK expression to promote EMT. We found that E2 increased MK mRNA expression in lung adenocarcinoma cells LTEP-a2 and A549 in a time-dependent manner. E2-induced MK expression was inhibited by the estrogen receptor (ER) antagonist ICI 182,780 and tamoxifen but not by phosphoinositide-3 kinase and MAPK inhibitors, suggesting a genomic mechanism of E2 on the regulation of MK transcription. Moreover, luciferase reporter and chromatin immunoprecipitation assays exhibited that E2 induced ERβ recruitment to the estrogen response element in the MK promoter. Small interfering RNA to ERα and ERβ revealed that ERβ mainly mediated E2-induced MK transcription. Interestingly, E2 enhanced MK expression in accordance with increase of EMT, whereas knockdown of MK could block EMT under E2 stimulation. Importantly, through analyzing lung adenocarcinoma tissues, there was indeed a correlation among levels of E2, MK, and EMT-related protein expression. Taken together, we reported a previously unrecognized mechanism on E2 in the regulation of MK expression and proved that MK plays a pivotal role in progression of E2-regulated EMT.
BACKGROUND:
The epithelial-to-mesenchymal transition (EMT) is a key process in carcinogenesis, invasion, and metastasis of oral squamous cell carcinoma (OSCC). In our previous studies, we found that neuropilin-1 (NRP1) is overexpressed in tongue squamous cell carcinoma and that this overexpression is associated with cell migration and invasion. Nuclear factor-kappa B (NF-κB) plays an essential role both in the induction and the maintenance of EMT and tumor metastasis. Therefore, we hypothesized that NRP1 induces EMT, and that NRP1-induced migration and invasion may be an important mechanism for promoting invasion and metastasis of OSCC through NF-κB activation.
METHODS/RESULTS:
The variations in gene and protein expression and the changes in the biological behavior of OSCC cell lines transfected with a vector encoding NRP1, or the corresponding vector control, were evaluated. NRP1 overexpression promoted EMT and was associated with enhanced invasive and metastatic properties. Furthermore, the induction of EMT promoted the acquisition of some cancer stem cell (CSC)-like characteristics in OSCC cells. We addressed whether selective inhibition of NF-κB suppresses the NRP1-mediated EMT by treating cells with pyrrolidinedithiocarbamate ammonium (PDTC), an inhibitor of NF-κB. Immunohistochemical analysis of NRP1 in OSCC tissue samples further supported a key mediator role for NRP1 in tumor progression, lymph node metastasis, and indicated that NRP1 is a predictor for poor prognosis in OSCC patients.
CONCLUSION:
Our results indicate that NRP1 may regulate the EMT process in OSCC cell lines through NF-κB activation, and that higher NRP1 expression levels are associated with lymph node metastasis and poor prognosis in OSCC patients. Further investigation of the role of NRP1 in tumorigenesis may help identify novel targets for the prevention and therapy of oral cancers.
BACKGROUND:
MiR-17-92 cluster and its paralogues have emerged as crucial regulators of many oncogenes and tumor suppressors. Transforming growth factor-β receptor II (TGFβR2), as an important tumor suppressor, is involved in various cancer types. However, it is in cancer that only two miRNAs of this cluster and its paralogues have been reported so far to regulate TGFβR2. MiR-93 is oncogenic, but its targetome in cancer has not been fully defined. The role of miR-93 in nasopharyngeal carcinoma (NPC) still remains largely unknown.
METHODS:
We firstly evaluated the clinical signature of TGFβR2 down-regulation in clinical samples, and next used a miRNA expression profiling analysis followed by multi-validations, including Luciferase reporter assay, to identify miRNAs targeting TGFβR2 in NPC. In vitro and in vivo studies were performed to further investigate the effects of miRNA-mediated TGFβR2 down-regulation on NPC aggressiveness. Finally, mechanism studies were conducted to explore the associated pathway and genes influenced by this miRNA-mediated TGFβR2 down-regulation.
RESULTS:
TGFβR2 was down-regulated in more than 50% of NPC patients. It is an unfavorable prognosis factor contributing to clinical NPC aggressiveness. A cluster set of 4 TGFβR2-associated miRNAs was identified; they are all from miR-17-92 cluster and its paralogues, of which miR-93 was one of the most significant miRNAs, directly targeting TGFβR2, promoting cell proliferation, invasion and metastasis in vitro and in vivo. Moreover, miR-93 resulted in the attenuation of Smad-dependent TGF-β signaling and the activation of PI3K/Akt pathway by suppressing TGFβR2, further promoting NPC cell uncontrolled growth, invasion, metastasis and EMT-like process. Impressively, the knockdown of TGFβR2 by siRNA displayed a consentaneous phenocopy with the effect of miR-93 in NPC cells, supporting TGFβR2 is a major target of miR-93. Our findings were also substantiated by investigation of the clinical signatures of miR-93 and TGFβR2 in NPC.
CONCLUSION:
The present study reports an involvement of miR-93-mediated TGFβR2 down-regulation in NPC aggressiveness, thus giving extended insights into molecular mechanisms underlying cancer aggressiveness. Approaches aimed at blocking miR-93 may serve as a promising therapeutic strategy for treating NPC patients.
AIM:
To investigate the mechanisms of how cyclooxygenase-2 (COX-2) regulates E-cadherin in gastric cancer cells.
METHODS:
COX-2 expression in human gastric cancer cell lines SGC-7901, BGC-823, MGC-803 and AGS were measured at the mRNA and protein level. COX-2 rich cell line SGC-7901 was chosen for subsequent experiments. siRNA mediated gene knockdown was used to investigate the impact of COX-2 on nuclear factor-κB (NF-κB), Snail, and E-cadherin in gastric cancer cells. Gene expression was determined by Western blot and real-time polymerase chain reaction. To analyze whether NF-κB inhibition could interrupt the modulatory effect of COX-2 or prostaglandin E2 (PGE2) on E-cadherin, gastric cancer cells were treated with celecoxib or PGE2, in the presence of NF-κB specific siRNA.
RESULTS:
Highest expression level of COX-2 was found in SGC-7901 cells, both at mRNA and protein levels. siRNA mediated down-regulation of COX-2 led to a reduced expression of NF-κB and Snail, but an increased expression of E-cadherin in SGC-7901 cells. siRNA mediated down-regulation of NF-κB also led to a reduced expression of E-cadherin and Snail in SGC-7901 cells. However, COX-2 expression did not alter after cells were treated with NF-κB specific siRNA in SGC-7901 cells. Treatment of SGC-7901 cells with celecoxib led to a reduced expression of Snail but an increased expression of E-cadherin. In contrast, treatment of SGC-7901 cells with PGE2 led to an increased Snail and a decreased E-cadherin. However, siRNA-mediated knockdown of NF-κB partially abolished the effect of celecoxib and PGE2 on the regulation of E-cadherin and Snail in SGC-7901 cells.
CONCLUSION:
COX-2 likely functions upstream of NF-κB and regulates the expression of E-cadherin via NF-κB/Snail signaling pathway in gastric cancer cells.
BACKGROUND:
Cancer stem cells (CSCs) are believed to be 'seed cell' in cancer recurrence and metastasis. MicroRNAs (miRNAs) can play an important role in the progression of primary tumor towards metastasis by regulating the epithelial-mesenchymal transition (EMT). The goal of this study was to investigate the effect of miRNA-200c overexpression on the EMT, tumorigenicity and metastasis of epithelial ovarian cancer (EOC) CSCs.
METHODS:
The EOC CD117+CD44+CSCs were isolated from the human ovarian cancer cell line SKOV3 by using a magnetic-activated cell sorting system, and the lentivirus miR-200c transduced CSCs were then selected for the study. The assays of colony forming, wound healing, cellular migration in vitro and tumor progression in vivo were performed.
RESULTS:
The miR-200c expression was reduced in the CD117+CD44+CSCs compared with the non-CD117+CD44+CSCs. However, the stable overexpression of the miR-200c in the CD117+CD44+CSCs resulted in a significant down-regulation of ZEB-1 and the Vimentin expression, an upregulation of the E-cadherin expression as well as a decrease of colony forming, migratory and invasion in vitro. Importantly, the miR-200c overexpression significantly inhibited the CD117+CD44+CSCs xenograft growth and lung metastasis in vivo in nude mice by inhibition of the EMT. In addition, the down-regulation of ZEB-1 showed the same efficacy as the miR-200c overexpression in the CD117+CD44+CSCs.
CONCLUSION:
These findings from this study suggest that the miR-200c overexpression may be considered a critical approach for the EOC CD117+CD44+CSCs in clinical trials.
AIM:
To investigate the significance of Twist2 for colorectal cancer (CRC).
METHODS:
In this study, 93 CRC patients were included who received curative surgery in Eastern Hepatobiliary Surgery Hospital from January 1999 to December 2010. Records of patients' clinicopathological characteristics and follow up data were reviewed. Formalin-fixed, paraffin-embedded tissue blocks were used to observe the protein expression of Twist2 and E-cadherin by immunohistochemistry. Two independent pathologists who were blinded to the clinical information performed semiquantitative scoring of immunostaining. A total score of 3-6 (sum of extent + intensity) was considered as Twist2-positive expression. The expression of E-cadherin was divided into two levels (preserved and reduced). An exploratory statistical analysis was conducted to determine the association between Twist2 expression and clinicopathological parameters, as well as E-cadherin expression. Furthermore, the variables associated with prognosis were analyzed by Cox's proportional hazards model. Kaplan-Meier analysis was used to plot survival curves according to different expression levels of Twist2.
RESULTS:
Twist2-positive expression was observed in 66 (71.0%) samples and mainly located in the cytoplasm. Forty-three (46.2%) samples showed reduced expression of E-cadherin. There were no significant correlations between Twist2 expression and any of the clinicopathological parameters. However, Twist2-positive expression was significantly associated with reduced expression of E-cadherin (P = 0.040). Multivariate analysis revealed that bad M-stage [hazard ratio (HR) = 7.694, 95%CI: 2.927-20.224, P < 0.001] and Twist2-positive (HR = 5.744, 95%CI: 1.347-24.298, P = 0.018) were the independent risk factors for poor overall survival (OS), while Twist2-positive (HR = 3.264, 95%CI: 1.455-7.375, P = 0.004), bad N-stage (HR = 2.149, 95%CI: 1.226-3.767, P = 0.008) and bad M-stage (HR = 10.907, 95%CI: 4.937-24.096, P < 0.001) were independently associated with poor disease-free survival (DFS). Survival curves showed a definite trend for Twist2-negative patients to have longer OS and DFS than Twist2-negative patients, not only overall, but also for patients in different stages, especially for DFS of patients in stage III (P = 0.033) and IV (P = 0.026).
CONCLUSION:
Our data suggests, for the first time, that Twist2 is a valuable prognostic biomarker for CRC, particularly for patients in stage III and IV.
Snail is closely linked to tumor invasion, metastasis, and recurrence and indicates prognosis of patients suffering from cancer. Overexpression of Snail increases motility and invasiveness of cancer cells, which has become target for anti-metastatic treatment. Oroxylin A, a natural compound extracted from Scutellaria radix, has been reported to inhibit invasion and migration in breast cancer. In this study, we investigated the anti-invasive effect of oroxylin A on lung cells and uncovered its underlying mechanism. The results suggested that oroxylin A could inhibit migration and invasion in Snail-expressing 95-D, and A549 cells whereas it had little effect on non-expressing GLC-82 cells. Furthermore, enhanced Snail expression after transfection of Snail vector in GLC-82 cells is decreased by oroxylin A. Snail can also induce epithelial-mesenchymal transition. We found oroxylin A could reverse TGFβ1-induced epithelial-mesenchymal transition by inhibiting Snail expression. As a result, oroxylin A up-regulated E-cadherin expression and down-regulated vimentin, MMP-9, and CD44v6 expression, which could lead to the inhibition of tumor migration and invasion. Mechanically, we demonstrated that oroxylin A suppressed activation of ERK instead of AKT pathway and then promoted activation of GSK-3β to reduce Snail protein content. Finally, we established transplanted, metastatic, and orthotopic models of A549 cells, and found that oroxylin A inhibited the growth and lung metastasis of A549 cells in vivo. Taken together, we proposed that oroxylin A might be a promising candidate targeting tumor metastasis.
© 2016 Wiley Periodicals, Inc.
Hepatocellular carcinoma (HCC) is a highly malignant tumor with an extremely poor prognosis. Our preliminary study indicated that bufalin could restrain the proliferation of human hepatoma BEL-7402 cells in a time- and dose-dependent manner. In the present study, the colony formation assay, the Transwell invasion assay, the western blot analysis and the immunofluorescence method were respectively used to investigate the effect and mechanism of bufalin against HCC cell invasion and metastasis. We found that: i) bufalin had significant inhibitory effect on the cell proliferation of BEL-7402 cells; ii) bufalin markedly inhibited the migration and invasion of BEL-7402 cells; iii) bufalin could suppress the phosphorylation of GSK-3β Ser9 site in BEL-7402 cells, decrease the expression of β-catenin, cyclin D1, metalloproteinases-7 (MMP-7) and cyclooxygenase-2 (COX-2) in the cytoplasm, and increase the expression of E-cadherin and β-catenin on the cell membrane; and iv) the expression of α-fetoprotein significantly decreased and the expression of albumin increased in BEL-7402 cells after bufalin was used. Our results indicate that: i) bufalin can regulate the expression of associated factors in Wnt/β-catenin signaling pathway of BEL-7402 cells through suppressing the phosphorylation of GSK-3β Ser9 site; ii) bufalin can strengthen intercellular E-cadherin/β-catenin complex to control epithelial-mesenchymal transition; and iii) bufalin can reverse the malignant phenotype and promote the differentiation and maturation by regulating the AFP and ALB expression in BEL-7402 cells. These are very important mechanisms of bufalin on the inhibition of the invasion and metastasis of HCC cells.
Downregulation of E-cadherin by the transcriptional repressor Snail is associated with acquisition of metastatic potential. Although hepatitis C virus (HCV) core protein has been implicated in hepatocarcinogenesis, it is unclear whether Snail is involved in HCV core-induced dysregulation of E-cadherin. Herein, we investigated the mechanism by which HCV core induces E-cadherin repression and the role of Snail in HCV core-mediated invasiveness and metastasis. We found that HCV infection, especially HCV core expression, effectively induced the epithelial-mesenchymal transition (EMT) in hepatoma cells by repressing E-cadherin. HCV core interacted with Snail and enhanced its binding to the E-box in the promoter region of E-cadherin, leading to decreased E-cadherin promoter activity. We found that HCV core, Snail, and the histone deacetylases HDAC1/HDAC2 formed a co-repressor complex at the E-cadherin promoter. Moreover, HCV core was shown to stabilize Snail through activation of the PI3K/Akt/GSK3β pathway. Silencing Snail expression restored E-cadherin expression and inhibited HCV core-promoted tumor growth and distant lung metastasis in vivo. Collectively, these results demonstrated that HCV core induced EMT by interacting with the transcriptional repressor complex Snail/HDACs at the E-cadherin promoter, which led to E-cadherin repression and increased invasiveness of hepatoma cells. These findings increase understanding of factors regulating metastasis in hepatoma and may ultimately lead to the development of novel treatment strategies for HCV-associated hepatocellular carcinoma.
Cancer related inflammation (CRI) is now recognized as the seventh hallmark in the pathogenesis of many types of malignancies. Paeonol, a natural phenolic component isolated from the root bark of Paeonia moutan, has significant anti-inflammatory activity. Recently, accumulating body of research has revealed potent anti-tumor effects mediated by paeonol. However, little is known about its anticancer mechanism on the basis of CRI. In this study, we observed that paeonol exerted direct anticancer activity through inhibition of cell proliferation, induction of apoptosis, and evident anti-inflammatory effects by reducing proinflammatory cytokines secretion (TNF-α, IL-1β, IL-6, and TGF-β) in the conditioned medium of B16F10 mouse melanoma cells. Interestingly, we found that paeonol significantly reversed motility phenotypes in TNF-α- or IL-6-induced B16F10 singe cell and collective migration and invasion in vitro, which were related to affecting epithelial-to-mesenchymal transition (EMT) makers and MMPs expression. In particular, paeonol disrupted both TNF-α-activated NF-κB and IL-6-activated STAT3 signaling pathways in B16F10 cells. EMSA and luciferase assays showed that paeonol abrogated NF-κB binding and NF-κB-driven promoter activity in the presence of TNF-α. Finally, we showed that paeonol attenuated B16F10 spontaneous lung metastases in C57/BL6J mice with down-regulated levels of serum proinflammatory cytokines. Therefore, paeonol possessed antitumor activity in melanoma cells and mice model by interruption of the aggressive feedback through proinflammatory cytokines mediated NF-κB and STAT3 signaling activation. These findings provide a novel treatment strategy that paeonol might be a promising versatile adjuvant therapy for cancer related inflammation.
Cancer stem cells (CSCs) are a subpopulation of neoplastic cells with self-renewal capacity and limitless proliferative potential as well as high invasion and migration capacity. These cells are commonly associated with epithelial-mesenchymal transition (EMT), which is also critical for tumor metastasis. Recent studies illustrate a direct link between EMT and stemness of cancer cells. Long non-coding RNAs (lncRNAs) have emerged as important new players in the regulation of multiple cellular processes in various diseases. To date, the role of lncRNAs in EMT-associated CSC stemness acquisition and maintenance remains unclear. In this study, we discovered that a set of lncRNAs were dysregulated in Twist-positive mammosphere cells using lncRNA microarray analysis. Multiple lncRNAs-associated canonical signaling pathways were identified via bioinformatics analysis. Especially, the Shh-GLI1 pathway associated lncRNA-Hh, transcriptionally regulated by Twist, directly targets GAS1 to stimulate the activation of hedgehog signaling (Hh). The activated Hh increases GLI1 expression, and enhances the expression of SOX2 and OCT4 to play a regulatory role in CSC maintenance. Thus, the mammosphere-formation efficiency (MFE) and the self-renewal capacity in vitro, and oncogenicity in vivo in Twist-positive breast cancer cells are elevated. lncRNA-Hh silence in Twist-positive breast cells attenuates the activated Shh-GLI1 signaling and decreases the CSC-associated SOX and OCT4 levels, thus reduces the MFE and tumorigenesis of transplanted tumor. Our results reveal that lncRNAs function as an important regulator endowing Twist-induced EMT cells to gain the CSC-like stemness properties.
Breast cancer is the most common cause of death among women. KIF3C, a member of kinesin superfamily, functions as a motor protein involved in axonal transport in neuronal cells. To explore the expression, regulation and mechanism of KIF3C in breast cancer, 4 breast cancer cell lines and 93 cases of primary breast cancer and paired adjacent tissues were examined. Immunohistochemistry, Real Time Polymerase Chain Reaction (RT-PCR), Western blot, flow cytometry, short hairpin RNA (shRNA) interference, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), colony formation techniques and xenograft mice model were used. We found that KIF3C was over-expressed in breast cancer tissues and such high KIF3C expression was also associated with tumor recurrence and lymph node metastasis. Silencing of KIF3C by shRNA inhibited epithelial-mesenchymal transition and metastasis by inhibiting TGF-β signaling and suppressed breast cancer cell proliferation through inducing G2/M phase arrest. The tumor size was smaller and the number of lung metastatic nodules was less in KIF3C depletion MDA-MB-231 cell xenograft mice than in negative control group. These results suggested that high expression of KIF3C in breast cancer may be associated with the tumor progression and metastasis.
The contribution of Ca(2+) in TGF-β-induced EMT is poorly understood. We aimed to confirm the effect of TGF-β on the gene expression of intracellular calcium-handling proteins and to investigate the potential underlying mechanisms in TGF-β-induced EMT. T47D and MCF-7 cells were cultured in vitro and treated with TGF-β. The mRNA expression of EMT marker genes and intracellular calcium-handling proteins were quantified by qRT-PCR. qRT-PCR and Western blot analysis results verified the changes of EMT marker gene expression. Furthermore, we found that TGF-β induced cell morphological changes significantly with an increase of cell surface area and cell length. These results indicated that TGF-β induced EMT. The mRNA expression levels of SPCA1, SPCA2 and MCU were not influenced by TGF-β treatment, while NCX1 expression was decreased in T47D cells. In addition, the mRNA levels of SERCAs and IP3Rs were significantly changed due to TGF-β-induced EMT. The TGF-β-treated T47D cells exhibited markedly greater response to ATP than the control cells, and the descent velocity of cytosolic calcium concentration was faster in TGF-β-treated cells than in control cells. This is the first report to demonstrate that TGF-β-induced EMT in human breast cancer cells is associated with alterations in endoplasmic reticulum calcium homeostasis.
Recent studies reveal that chemotherapy can enhance metastasis due to host responses, such as augmented expression of adhesion molecules in endothelial cells and increased populations of myeloid cells. However, it is still unclear how tumour cells contribute to this process. Here, we observed that paclitaxel and carboplatin accelerated lung metastasis in tumour-bearing mice, while doxorubicin and fluorouracil did not. Mechanistically, paclitaxel and carboplatin induced similar changes in cytokine and angiogenic factors. Increased levels of CXCR2, CXCR4, S1P/S1PR1, PlGF and PDGF-BB were identified in the serum or primary tumour tissues of tumour-bearing mice treated by paclitaxel. The serum levels of CXCL1 and PDGF-BB and the tissue level of CXCR4 were also elevated by carboplatin. On the other hand, doxorubicin and fluorouracil did not induce such changes. The chemotherapy-induced cytokine and angiogenic factor changes were also confirmed in gene expression datasets from human patients following chemotherapy treatment. These chemotherapy-enhanced cytokines and angiogenic factors further induced angiogenesis, destabilized vascular integrity, recruited BMDCs to metastatic organs and mediated the proliferation, migration and epithelial-to-mesenchymal transition of tumour cells. Interestingly, inhibitors of these factors counteracted chemotherapy-enhanced metastasis in both tumour-bearing mice and normal mice injected intravenously with B16F10-GFP cells. In particular, blockade of the SDF-1α-CXCR4 or S1P-S1PR1 axes not only compromised chemotherapy-induced metastasis but also prolonged the median survival time by 33.9% and 40.3%, respectively. The current study delineates the mechanism of chemotherapy-induced metastasis and provides novel therapeutic strategies to counterbalance pro-metastatic effects of chemo-drugs via combination treatment with anti-cytokine/anti-angiogenic therapy.
TGF-β1 secreted abundantly by tumors cells as well as present in the local microenvironment promotes neoplasm invasion and metastasis by triggering the epithelial to mesenchymal transition (EMT). MiR200c has been shown to suppress EMT and to regulate the cellular epithelial and interstitial state conversion, whereas the tumor vaccines are intended to specifically initiate or amplify a host response against evolving tumor cells. Our study aimed at optimizing the antitumor effects of the B16F10/glycosylphosphatidylinositol-interleukin 21 (B16F10/GPI-IL-21) tumor vaccine on melanoma bearing mice by combining the TGF-β1 knockdown and the administration of miR200c agomir. The mice were subcutaneously vaccinated with inactivated B16F10/GPI-IL-21 vaccine and challenged by B16F10 cells transfected with shTGF-β1 (B16F10/shTGF-β1 cells) or B16F10/shTGF-β1 cells with the administration of miR200c agomir. The later combination showed that, when compared with the mice in the control group that received no vaccination, vaccinated mice significantly increased NK and CTL activities, enhanced levels of IFN-γ, and reduced expression of TGF-β1, N-cadherin, Vimentin, Gli1/2, P-Smad2/3 and others involved in promoting expression of EMT-related molecules in tumor areas, and inhibited the melanoma metastasis in lungs and lymph nodes. Altogether, our findings demonstrate that this synergistic anti-cancer regimen effectively induces strong immune response and diminishes the melanoma progression.
Hairy enhancer of split-1 (HES1) is a transcriptional target of the Notch pathway, and a high level of HES1 is regarded as a marker of activated Notch. The aim of the study was to investigate the role of HES1 in colorectal cancer progression. We used tissue microarrays to analyze the expression and clinical significance of HES1 in 320 colorectal cancer samples. Stable overexpression and knockdown of HES1 were established in three colorectal cancer cell (CRC) lines (RKO, HCT8 and LOVO). We investigated the differentially expressed genes and enriched pathways in HES1 overexpressing CRC cells by gene expression profiling. Also, the role of HES1 in invasion and migration were examined in vitro and in vivo. We found that high expression of HES1 was significantly correlated with distal metastasis (P = 0.037) at diagnosis, and HES1 could serve as an unfavorable prognostic factor for colorectal cancer patients (P = 0.034). Gene expression profiling and pathway enrichment analysis revealed that HES1 was related to cellular adherens junction loss. In addition, we showed that HES1 overexpression lead to depressed E-cadherin, and elevated N-cadherin, vimentin and Twist-1 levels. Functionally, HES1 enhanced invasiveness and metastasis of CRC cells. HES1 promotes cancer metastasis via inducing epithelial mesenchymal transition and serves as a poor prognosis factor of colorectal cancer patients.
Metastasis is the leading cause of death in lung cancer. Understanding the mechanisms underlying the process of metastasis is crucial for identifying novel anti-metastatic therapies. Studies indicate that the highly conserved developmental pathways, such as the Wnt and Notch signaling pathways, play important roles in the non-small cell lung cancer (NSCLC) tumorigenesis. However, the roles of both pathways in NSCLC metastasis are unclear. The present study aimed to investigate whether Wnt3a and Notch3, key components of the Wnt and Notch signaling pathways, respectively, regulate the metastatic abilities of NSCLC cells and whether there is some relationship during these regulatory events. Here, we observed that Wnt3a treatment upregulated, not only the protein expression of Notch3, but also the mRNA expression of Notch3 and its downstream genes, HES1 and HEYL. In addition, Wnt3a promoted cell invasion and anchorage-independent growth. Meanwhile, Wnt3a treatment caused epithelial‑mesenchymal transition (EMT)-like morphological changes and F-actin reorganization. The western blotting data showed that Wnt3a treatment decreased the expression of E-cadherin and increased the expression of N-cadherin and vimentin. Compared with Wnt3a treatment, Notch3 shRNA transfection had opposite effects. Furthermore, Notch3 shRNA weakened the effects of Wnt3a treatment on the in vitro cell invasion and EMT. Overall, these observations suggest that Wnt3a and Notch3 may promote the metastasis of NSCLC and Notch3 upregulation is required for the Wnt3a mediated increased metastatic abilities of NSCLC.
The effect of proton pump inhibitor (PPI) on cancer risk has received much attention recently. In this study, we investigated the mechanism underlying multidrug resistance and the effect of a PPI pantoprazole using an adriamycin-resistant gastric cancer cell model (SGC7901/ADR). Compared with the parental cell line, SGC7901/ADR cells showed reduced proliferation rate, but higher resistance to adriamycin under both anchorage-dependent and -independent conditions. Notably, SGC7901/ADR cells underwent epithelial to mesenchymal transition (EMT) and showed increased migrating and invading capabilities. At molecular level, SGC7901/ADR cells showed strong activation of Wnt/β-catenin signaling pathway compared with parental sensitive cells. Interestingly, we found that a PPI pantoprazole can effectively reverse the aggressiveness and EMT marker expression of SGC7901/ADR cells. Furthermore, pantoprazole treatment resulted in a profound reduction of both total and phosphorylated forms of Akt and GSK-3β, which in turn suppressed the adriamycin-induced Wnt/β-catenin signaling in SGC7901/ADR cells. Taken together, we demonstrate that the aggressive phenotype of adriamycin-resistant SGC7901/ADR cells is mediated by induction of EMT and activation of the canonical Wnt/β-catenin signaling pathway. And for the first time, we show that it is possible to suppress the invasiveness of SGC7901/ADR cells by pantoprazole which targets the EMT and Akt/GSK-3β/β-catenin signaling.
Our previous works showed chronic infection of Mycoplasma hyorhinis (M. hyorhinis) was associated with gastric cancer metastasis, but the mechanisms were unknown. Herein, we found M. hyorhinis induced epithelial-mesenchymal transition (EMT) in gastric cancer cell MGC803, which was counteracted by inhibitor of NF-κB signaling or p65 knockdown. Furthermore, we found that TLR4 associated with p37, a membrane protein of M. hyorhinis. Knock-down or inhibition of TLR4 antagonized M. hyorhinis-induced NF-κB signaling, EMT, and cell migration. Thus, M. hyorhinis induces EMT and promotes cell migration via TLR4-NF-κB signaling, which provides a clue to the pathogenesis of M. hyorhinis in gastric cancer.
Accumulating evidences implicate that ribonuclease inhibitor (RI) plays a suppressing role in cancer development. However, the mechanisms underlying antitumor of RI remain largely unknown. Epithelial-mesenchymal transition (EMT) is regarded as a key event in tumor progression. The reports have demonstrated that EMT was implicated in metastasis of bladder cancer. Therefore, we suppose that RI might involve regulating EMT of bladder cancer. Here bladder cancer T24 cells were transfected with pGensil-1-siRNA-RI vectors. HE staining, living cell observation, Phalloidine-FITC staining of microfilament, cell adhesion, scratch migration, and Matrigel invasion were examined respectively. RI expression and colocalization with ILK were detected using confocal microscope. Proteins associated with EMT were determined with Western blotting and immunohistochemistry in vivo and in vitro. Effects of RI expression on tumor growth, metastasis and EMT related proteins in BALB/C nude mouse and clinical human bladder cancer specimens were valued with histological, immunohistochemical and immunofluorescent examination respectively. We demonstrated that down-regulating RI increased cell proliferation, migration and invasion, changed cell morphology and adhesion, and rearranged cytoskeleton by inducing EMT and ILK signaling pathway in bladder cancer cells. In addition, we showed that down-regulating RI promoted tumorigenesis and metastasis of bladder cancer in vivo. Finally, we found that bladder cancer with invasive capability had higher Vimentin, Snail, Slug and Twist as well as lower E-cadherin and RI expression in clinical human specimens. Our results suggest that RI could play a novel role in inhibiting metastasis of bladder through regulating EMT and ILK signaling pathway.
Expression of epithelial-mesenchymal transition (EMT) markers has been detected clinically in benign prostatic hyperplasia (BPH) tissues. To understand the molecular basis, we investigated the role of stromal microenvironment in the progression of EMT in BPH cells. First, we used cell culture supernatant from normal prostate stromal WPMY-1 cells to provide supernatant-conditioned medium (WSCM) to culture the BPH-1 cell line. Then, the morphological changes and migratory capacity were detected in BPH-1 cells. The expression of EMT markers was examined in BPH-1 cells by Western blot and immunofluorescent analysis. Finally, to investigate the role of transforming growth factor beta 1 (TGF-β1) in this process, the WSCM-cultured cells were treated with monoclonal antibody against TGF-β1 to study its effect on EMT. We found that the morphology of BPH-1 cells changed to a spindle-like shape after cultured in WSCM, and the levels of E-cadherin and cytokeratin 5/8 (CK5/8) were significantly lower than the cells cultured in ordinary medium. These BPH-1 cells were also tested positive for mesenchymal markers vimentin and a-smooth muscle actin (SMA) as well as Snail. We also found WSCM can increase the migratory capacity of BPH-1 cells. In addition, when they were treated with anti-TGF-β1, upregulation of E-cadherin and CK5/8 levels was observed but no expression of vimentin, alpha-SMA or Snail was detected. Furthermore, phosphorylated-Smad3 expression in WSCM-cultured BPH-1 cells was also suppressed by anti-TGF-β1 treatment. Our results demonstrated that stromal cell supernatant was able to induce EMT in BPH-1 cells, possibly through secreting TGF-β1 to activate Smad signaling. Our results suggest novel molecular targets for clinical treatment of BPH by modification of stromal microenvironment through inhibiting TGF-β1/Smad expression.
Tumor invasion and migration obstructs the treatment and prognosis of cancer. In this research, we investigated the effect of oroxylin A, a natural compound extracted from Scutellaria radix, the root of Scutellaria baicalensis, on inhibition of the invasion and migration of three different tumor cell lines: MCF-7, DU145, and HepG2. The results suggested that oroxylin A could inhibit hypoxia-induced migration and invasion of the three cell lines mentioned above. To study the detailed mechanisms, studies were carried out on MCF-7 cells and it was found that oroxylin A could regulate the expression of related markers in MCF-7 cells including E-cadherin, N-cadherin, and Vimentin. It was also found that oroxylin A inhibited the hypoxia-induced invasion and migration of MCF-7 cells by suppressing the Notch pathway. Oroxylin A inhibited N1ICD translocating to the nucleus and binding to epithelial-mesenchymal transition-related transcription factor Snail, thus suppressing the invasion and migration of MCF-7 cells. Therefore, oroxylin A is expected to be a promising candidate for antimetastasis treatment through suppression of the hypoxia-induced Notch pathway.
Secreted frizzled-related proteins (SFRPs) are antagonists of the Wnt signaling pathway whose epigenetic downregulation have been shown to be involved in hepatocarcinogenesis. However, dysregulation of SFRPs induced by hepatitis B virus (HBV) X protein (HBx) has never been studied in HBV-related hepatocellular carcinoma (HBV-HCC). In this study, we sought to determine the clinical significance and underlying mechanism of HBx-induced SFRPs dysregulation in hepatoma cells and HBV-HCC patients. Our results showed that SFRP1 and SFRP5 expression were dramatically decreased by HBx in hepatoma cells. The repressed expression in hepatoma cells was partially rescued by a DNA methylation inhibitor and synergistically increased by a combination treatment with a histone deacetyltransferases inhibitor. In addition, we identified that SFRP1 and SFRP5 promoters were hypermethylated in both HBx-expressing hepatoma cells and HBV-HCC tissues. Downregulation of SFRP1 and SFRP5 in HBV-HCC tissues was significantly correlated with overexpression of DNA methyltransferase 1 (DNMT1) and poor tumor differentiation. HBx facilitated the binding of DNMT1 and DNMT3A to SFRP1 and SFRP5 promoters, and resulted in epigenetic silencing of SFRP1 and SFRP5. Moreover, overexpression of SFRP1, SFRP5 or RNA interference mediated silencing of DNMT1 inactivated the Wnt signaling pathway and decreased the expression levels of Wnt target genes c-Myc and CyclinD1, thus impeding HCC growth in vitro and in vivo, and regressing HBx-induced epithelial-mesenchymal transition (EMT). Our findings strongly suggest that epigenetic silencing of SFRP1 and SFRP5 by HBx allows constitutive activation of Wnt signaling pathway and hence contributes to hepatocarcinogenesis.
Mesenchymal stem cell (MSC) tropism to injured tissue sites in response to inflammation and wounds has been suggested. MSC activation and recruitment by Helicobacter pylori (H. pylori)-infected gastrointestinal epithelial cells has been demonstrated. As a component of the chronic gastritis microenvironment, MSCs play critical roles in the development of H. pylori-associated gastric mucosal lesions/malignancies. However, the mechanisms responsible for this process remain largely unknown. In this study, we demonstrate that H. pylori infection induces the differentiation of MSCs into cancer-associated fibroblast (CAF)-like cells. H. pylori-infected MSCs possessed an altered cytokine profile and induced epithelial-mesenchymal transition in gastric epithelial cells, leading to destroyed cell junctions, enhanced cell migration, reduced cell apoptosis and increased oncogenic potential. In conclusion, our findings indicate that H. pylori infection may cause gastric lesions/malignancies by inducing the differentiation of MSCs into CAFs and suggest a novel mechanism of action and role of MSCs in the development and progression of gastric cancer.
The epithelial-to-mesenchymal transient (EMT) is associated with tumor metastasis. Twist is one of the key transcription factors for EMT and relates to tumor cell migration. Long non-coding RNAs (lncRNAs) have recently emerged as important regulatory molecules involved in a broad range of biological processes and complicated diseases. However, it is unknown whether a signal network and lncRNAs are involved in Twist-induced EMT program. Taking MCF10A/Twist as a model, more than 99 lncRNAs and 3164 genes are regulated in the Twist-induced EMT process using lncRNA-array and cDNA micro-array. We establish a downstream signal network associated with EMT induced by Twist using bioinformatic analysis (Gene Ontology, pathway analysis) and experimental data. A set of multiple canonical signal pathways (such as WNT, MAPK, JAK/STAT, TGF-β, mTOR, Hedgehog and P53 signaling pathways) and several lncRNAs [such as lncRNA (chr6, 26124411-26139312, +), lncRNA (chr1, 41944445-41949874, -), lncRNA (chr17, 44833874-44834830, +)] are altered in MCF10A/Twist cells. More interestingly, lncRNA (chr17, 44833874-44834830, +), lncRNA (chr17, 21142183-21156578, -), lncRNA (chr6, 26124411-26139312, +) and lncRNA (chr19, 438420-2083745, -) may be involved in regulation or activation of WNT signaling pathway in the Twist-induced EMT process. These findings first determine that Twist contributes to invasion and metastasis by inducing wide-ranging transcriptional and functional changes of lncRNAs and signal pathways in our study.
microRNAs (miRNAs) play essential roles in several physiological and pathological processes, including tumor metastasis. Metastasis is associated with poor prognosis in renal carcinoma patients and almost 20-30% of patients present with distant metastasis at the time of diagnosis. The aim of the present study was to investigate the possible roles of miR-200c in regulating metastasis and to identify its target genes in renal cell carcinoma (RCC). Among the miRNAs downregulated in our tissue specimen microarray, miR-200c was downregulated significantly. Functional assays demonstrated that restoration of miR-200c significantly inhibited the migration and invasion of SN12-PM6 and 786-0 cells in vitro. Genome-wide gene expression analysis and TargetScan database studies showed that ZEB1, which has been shown to promote tumor invasion and migration through E-cadherin gene silencing, is a promising candidate target gene of miR‑200c. Overexpression of miR-200c in SN12-PM6 and 786-0 cells was concurrent with downregulation of ZEB1 and upregulation of E-cadherin mRNA and protein. In addition, miR-200c affected the protein expression of p-Akt and Akt. Thus, our study demonstrated that miR-200c decreases the metastatic ability of renal carcinoma cells by upregulating E-cadherin through ZEB1 and that modulating the expression of miR-200c could influence Akt protein levels. We therefore concluded that there is an Akt-miR-200c-E-cadherin axis in the epithelial-to-mesenchymal transition process in RCC.
Integrin-linked kinase (ILK) is a multifunctional serine/threonine kinase. Accumulating evidences suggest that ILK are involved in cell-matrix interactions, cell proliferation, invasion, migration, angiogenesis and Epithelial-mesenchymal transition (EMT). However, the underlying mechanisms remain largely unknown. EMT has been postulated as a prerequisite for metastasis. The reports have demonstrated that EMT was implicated in metastasis of oral squamous cell carcinomas. Therefore, here we further postulate that ILK might participate in EMT of tongue cancer. We showed that ILK siRNA inhibited EMT with low N-cadherin, Vimentin, Snail, Slug and Twist as well as high E-cadherin expression in vivo and in vitro. We found that knockdown of ILK inhibited cell proliferation, migration and invasion as well as changed cell morphology. We also demonstrated that ILK siRNA inhibited phosphorylation of downstream signaling targets Akt and GSK3β as well as reduced expression of MMP2 and MMP9. Furthermore, we found that the tongue tumor with high metastasis capability showed higher ILK, Vimentin, Snail, Slug and Twist as well as lower E-cadherin expression in clinical specimens. Finally, ILK siRNA led to the suppression for tumorigenesis and metastasis in vivo. Our findings suggest that ILK could be a novel diagnostic and therapeutic target for tongue cancer.
Human ribonuclease inhibitor (RI) is a cytoplasmic acidic protein possibly involved in biological functions other than the inhibition of RNase A and angiogenin activities. We have previously shown that RI can inhibit growth and metastasis in some cancer cells. Epithelial-mesenchymal transition (EMT) is regarded as the beginning of invasion and metastasis and has been implicated in the metastasis of bladder cancer. We therefore postulate that RI regulates EMT of bladder cancer cells. We find that the over-expression of RI induces the up-regulation of E-cadherin, accompanied with the decreased expression of proteins associated with EMT, such as N-cadherin, Snail, Slug, vimentin and Twist and of matrix metalloprotein-2 (MMP-2), MMP-9 and Cyclin-D1, both in vitro and in vivo. The up-regulation of RI inhibits cell proliferation, migration and invasion, alters cell morphology and adhesion and leads to the rearrangement of the cytoskeleton in vitro. We also demonstrate that the up-regulation of RI can decrease the expression of integrin-linked kinase (ILK), a central component of signaling cascades controlling an array of biological processes. The over-expression of RI reduces the phosphorylation of the ILK downstream signaling targets p-Akt and p-GSK3β in T24 cells. We further find that bladder cancer with a high-metastasis capability shows higher vimentin, Snail, Slug and Twist and lower E-cadherin and RI expression in human clinical specimens. Finally, we provide evidence that the up-regulation of RI inhibits tumorigenesis and metastasis of bladder cancer in vivo. Thus, RI might play a novel role in the development of bladder cancer through regulating EMT and the ILK signaling pathway.
MicroRNAs (miRNAs) have been believed to associate with malignant progression including cancer cell proliferation, apoptosis, differentiation, angiogenesis, invasion and metastasis. However, the functions of miRNAs are intricate, one miRNA can directly or indirectly target multiple genes and function as oncogene or tumor suppressor gene. In this study, we found that miR-21 inhibits PTEN and human sulfatase-1 (hSulf-1) expression in hepatocellular carcinoma (HCC) cells. The hSulf-1 is a heparin-degrading endosulfatase, which can inhibit the heparin binding growth factor-mediated signaling transduction into cells. Therefore, miR-21-mediated suppression of both hSulf-1 and PTEN led to activation of AKT/ERK pathways and epithelial-mesenchymal transition (EMT) in HCC cells, and finally enhance the activity of HCC cell proliferation and movement and promote HCC xenograft tumor growth in mouse models. These findings may provide candidate targets for prevention and treatment of HCC.
The inhibitor of apoptosis proteins (IAP) are closely correlated with proliferation, apoptosis, motility, and metastasis. Livin is the most recently identified IAP, and its role in breast progression remains unknown. In our study, analyses of 50 patients with breast cancer revealed that the positive expression rate of Livin was higher in breast cancer tissues (62%) relative to that in adjacent (35%) and normal tissues (25%). Livin expression in breast cancer correlated with the clinical stage and axillary lymph node metastasis and could be used as a prognostic marker. Our in vitro experiment revealed that Livin was highly expressed in high-invasive MDA-MB-231 cells as compared to low-invasive cells (MCF-7). Suppression of Livin by short-hairpin RNA reduced the Livin expression of MDA-MB-231 cells and subsequently inhibited tumor cell growth, proliferation, and colony formation and induced tumor cell apoptosis, motility, migration, and invasion. Overexpression of Livin in MCF7 cells resulted in increased migration and invasion capabilities of the cells without affecting proliferation and apoptosis. In addition, epithelial-mesenchymal transition (EMT) was induced by Livin expression in breast cancer cell lines. The high level of phosphorylated AKT in MDA-MB-231 cells was suppressed by Livin knockdown. Further, Livin-induced migration and invasion could be abolished by either the application of the phosphoinositide-3-kinase inhibitor LY294002 or knockdown of AKT expression using small-interfering RNA. In conclusion, Livin serves as an independent prognostic indicator for breast cancer. Livin expression promotes breast cancer metastasis through the activation of AKT signaling and induction of EMT in breast cancer cells both in vitro and in vivo.
This study was designed to investigate the putative protective effect of β-casomorphin-7 on diabetic nephropathy in a rat model, and to explore the possible mechanism of this effect. SD rats were randomly divided into the following three groups: control group, diabetes group and β-casomorphin-7-treatment group. All rats were euthanized after 30 days with or without β-casomorphin-7 treatment. Biochemical parameters including blood glucose and renal function were quantified. The concentration of plasma TGF-β1 was measured by ELISA. Histopathological changes to the kidney were studied by Masson and Sirius red staining. Expressions of α-smooth muscle actin (α-SMA), E-cadherin, vimentin, cytokeratin19 and TGF-β1 mRNA in rat renal cortices were analyzed by real-time PCR. Changes in α-SMA and E-cadherin protein expression in rat renal cortices were quantified by Western blot. β-Casomorphin-7 treatment of diabetic rats reduced urinary glucose, urinary protein, serum creatinine, blood urinary nitrogen, plasma TGF-β1 and the ratio of kidney: body weight. Masson and Sirius red staining showed that β-casomorphin-7 treatment attenuated renal interstitial fibrosis in diabetic rats. Compared to the control rats, diabetic rats had elevated expressions of α-SMA, vimentin and TGF-β1 mRNA and α -SMA protein and decreased expression of E-cadherin and cytokeratin19 mRNA, and E-cadherin protein. β-Casomorphin-7 treatment of diabetic rats partially normalized these changes. Our results suggest that administration of β-casomorphin-7 attenuates renal interstitial fibrosis caused by diabetes. This protective effect may be associated, in part, with down regulation of epithelial-mesenchymal transition of renal tubular epithelial cells.
Integrin-linked kinase (ILK) is a multifunctional serine/threonine kinase in cytoplasm. Recent studies showed that cancer patients with increased ILK expression had low survival, poor prognosis and increased metastasis. Although the causes of ILK overexpression remain to be fully elucidated, accumulating evidence suggests that its oncogenic capacity derives from its regulation of several downstream targets that provide cells with signals that promote proliferation, survival and migration. However, the mechanisms underlying tumor metastasis by ILK is still not fully understood. Epithelial–mesenchymal transition (EMT) is a critical event of cancer cells that triggers invasion and metastasis. We recently reported that knockdown of ILK inhibited the growth and induced apoptosis in human bladder cancer cells. Therefore, we postulate that ILK might involve in EMT. Here we further investigate the function of ILK with RNA interference in bladder cancer cells. Knockdown of ILK impeded an EMT with low Vimentin, Snail, Slug and Twist as well as high E-cadherin expression in vivo and vitro. In addition, we found that knockdown of ILK inhibited cell proliferation, migration and invasion as well as changed cell morphology, adhesion and rearranged cytoskeleton in vitro. We also demonstrated that ILK siRNA inhibited phosphorylation of downstream signaling targets Akt and GSK3β, increased expression of nm23-H1, as well as reduced expression of MMP-2 and MMP-9 in vivo and vitro. Furthermore, downregulation of ILK could increase expression of Ribonuclease inhibitor (RI), an important acidic cytoplasmic protein with many functions. Finally, the effects of ILK siRNA on bladder cancer cell phenotype and invasiveness translate into suppression for tumorigenesis and metastasis in vivo. Taken together, our findings highlight that ILK signaling pathway plays a novel role in the development of bladder cancer through regulating EMT. ILK could be a promising diagnostic marker and therapeutic target for bladder cancer.
Human ribonuclease inhibitor (RI) is a cytoplasmic acidic protein. RI is constructed almost entirely of leucine rich repeats, which might be involved in unknown biological effects except inhibiting RNase A and angiogenin activities. We previously reported that up-regulating RI inhibited the growth and metastasis of melanoma cells. Epithelial-mesenchymal transition (EMT) is a critical event of cancer cells that triggers invasion and metastasis. However, the role of RI in the EMT process remains unknown. Here we hypothesize that RI might inhibit melanoma invasion and metastasis by regulating EMT. We found that over-expression of RI induced up-regulation of E-cadherin, accompanied with decreased expressions of proteins associated with EMT such as N-cadherin, Snail, Slug, Vimentin and Twist both in vitro and in vivo. Furthermore, RI restrained matrix metalloproteinase MMP-2 and MMP-9 secretions in B16 and B16-F10 melanoma cells. In addition, we also found that up-regulation of RI inhibited cell proliferation, migration and invasion as well as changed cell morphology, adhesion and rearranged cytoskeleton in vitro. Finally, the effects of RI on phenotype and invasiveness translated into suppressing metastasis by the experimental metastasis models of melanoma with lighter lung weight, a fewer metastasis nodules and a lower incidence rate, with respect to the control groups. Taken together, our data highlight, for the first time, that RI plays a novel role in inhibiting development and progression of murine melanoma cells through regulating EMT. These results suggest that RI could be a therapeutic target protein for melanoma and may be of biological importance.
Osteopontin (OPN) has been implicated in the pathology of several renal conditions. Recently, we demonstrated in vitro that aldosterone has important roles in collagen synthesis by inducing OPN (Irita J, Okura T, Kurata M, Miyoshi K, Fukuoka T, Higaki J. Hypertension 51: 507-513, 2008). The aim of the present study was to clarify the roles of OPN in aldosterone-mediated renal fibrosis by infusing aldosterone into either wild-type (WT) or OPN knockout mice (OPN(-/-)). We used uninephrectomized mice treated with aldosterone and high salt to exacerbate renal fibrosis. After 4 wk of treatment with aldosterone, we showed similar increases in systolic blood pressure in both strains of mice. Urine albumin excretion was greater in aldosterone-infused WT mice than in aldosterone-infused OPN(-/-) mice. Immunohistochemical analysis showed high levels of OPN expression in aldosterone-infused WT mice. Interstitial fibrosis and inflammatory infiltrations were increased in aldosterone-infused WT mice compared with either vehicle-infused WT or aldosterone-infused OPN(-/-) mice. These changes were ameliorated markedly by eplerenone treatment in aldosterone-infused WT mice. Aldosterone-infused WT mice also had increased expression of NADPH oxidase subunits compared with aldosterone-infused OPN(-/-) mice. We observed a marked increase in oxidative stress markers in aldosterone-infused WT mice compared with aldosterone-infused OPN(-/-) mice. These results indicate that OPN is a promoter of aldosterone-induced inflammation, oxidative stress, and interstitial fibrosis in the kidney and suggest that inhibition of OPN may be a potential therapeutic target for prevention of renal injury.
Human ribonuclease inhibitor (RI) is a cytoplasmic acidic protein. RI is constructed almost entirely of leucine-rich repeats, which might be involved in some unknown biological functions like other structurally similar proteins besides inhibiting RNase A and angiogenin activities. Our previous experiments demonstrated that up-regulating RI might effectively inhibit some tumor growth and metastasis. However, the down-regulating RI influence on the tumor does not have any report until now, the mechanisms underlying antitumor of RI have not been fully understood. In this study, the efficient RNA interferences of RI were constructed using a plasmid vector and identified with RT-PCR, Western blot and Immunocytochemistry, then were transfected into non-invasive bladder cancer BIU-87 cells. We demonstrated that knockdown RI expression in BIU-87 cells could obviously change the cell morphology, rearrange the microfilaments and extend the lamellipodia, as well as enhance proliferation, increase migration, invasion and matrix metalloprotease level, and also reduce adhesion in vitro. BALB/C nude mice that were injected with the BIU-87 cells transfected RI siRNA showed a significant facilitation of the tumor with heavier tumor weight, higher density of microvessels, lower nm23-H1 and E-Cadherin expressions than those in the control group. Taken together, these experiments suggest that knockdown of RI could promote growth and metastasis potentials of BIU-87 cells. Our present findings reveal the novel mechanism that anti-tumor effect of RI is also involved in suppressing growth and metastasis, besides antiangiogenesis. The results show that RI may be a therapeutic target protein for bladder cancer and may be of biological importance.
To investigate the influence of fasudil on the epithelial-mesenchymal transdifferentiation of renal tubular epithelial cells from diabetic rats and explore the mechanisms of this effect. Wistar rats were randomly divided into the following three groups: control, diabetes and fasudil-treatment. All rats were sacrificed after three months of feeding with or without fasudil treatment. Pathological changes to the glomeruli and renal interstitium were studied using Periodic acid-Schiff's staining and Masson staining, respectively. Expression of ROCK1, alpha-SMA, E-cadherin and the distribution of beta-catenin in rat renal cortex were revealed by immunohistochemistry. Changes in the MYPT1 phosphorylation profile and alpha-SMA, E-cadherin and membrane beta-catenin expression were revealed by western blot. Changes in the levels of ROCK1, E-cadherin and total beta-catenin mRNA expression were analyzed by real-time PCR. Fasudil treatment notably attenuates renal interstitial fibrosis in diabetic rats. Compared to the control rats, diabetic rats showed elevated phosphorylation of MYPT1, increased expression of ROCK1 and alpha-SMA, decreased expression of E-cadherin and membrane beta-catenin, and increased expression of ROCK1 and total beta-catenin mRNA, decreased expression of E-cadherin mRNA. Fasudil treatment of diabetic rats resulted in attenuated MYPT1 phosphorylation, decreased ROCK1 and alpha-SMA expression, increased E-cadherin and membrane beta-catenin expression, and reduced ROCK1 and total beta-catenin mRNA expression, increased expression of E-cadherin mRNA. In conclusion, fasudil may reduce the epithelial-mesenchymal transdifferentiation and renal interstitial fibrosis in diabetic rats through a mechanism by which ROCK activity is inhibited, which further facilitates the recovery of the cell-cell adhesions among renal tubular epithelial cells and adhesion complex formation.
BACKGROUND:
The gastric dramatic down-related gene (GDDR) is an abundantly expressed secretory protein in normal gastric epithelia, while its expression is distinctly decreased in gastric cancer. However, the role of GDDR in gastric cancer remains poorly understood.
AIMS:
This study aims to detect the expression and clinical significance of GDDR in gastric cancer and investigate its effects on epithelial-mesenchymal transition.
METHODS:
The expression of GDDR in gastric cancer was examined by immunohistochemistry, immunoblotting, and Western blotting. The relationships between GDDR expression and clinicopathological factors were evaluated. The effects of GDDR on epithelial-mesenchymal transition of gastric cancer cells were investigated in vitro.
RESULTS:
GDDR was absent in gastric cancer tissue or dramatically downregulated in gastric cancer cell lines. Loss of GDDR expression in gastric cancer was strongly correlated with clinicopathological factors, such as tumor differentiation (p = 0.037), T stage (p < 0.001), lymph node metastasis (p = 0.008) and TNM stage (p < 0.001). Patients with decreased GDDR expression presented shortened overall survival (p = 0.033). Functional studies demonstrated that GDDR elevation augmented cell-cell adhesion and suppressed cell motility, concomitant with increased expression of E-cadherin and decreased expression of β-catenin and vimentin. Conversely, GDDR depletion increased cell motility, concomitant with decreased expression of E-cadherin and increased expression of β-catenin and vimentin. Moreover, GDDR had an inhibitory effect on PI3K/Akt signaling pathway.
CONCLUSIONS:
Our findings suggested that GDDR expression was significantly associated with the progression of gastric cancer and GDDR may function as a tumor suppressor via inhibiting the epithelial-mesenchymal transition.
BACKGROUND:
The cAMP-PKA signaling pathway and TGF-β1-dependent fibrosis pathways are of particular importance in ADPKD progression, but the cross-talk between these pathways remains unclear. Therefore, we used an MDCK-cell model and embryonic kidney-cyst model to study the regulatory role of cAMP-PKA signaling in the TGF-β1 induced fibrotic process.
METHOD AND RESULTS:
Pkd1(flox/flox); Ksp-Cre and Pkd1(+/+); Ksp-Cre mice were used as an in vivo model. Increased kidney volume, renal cysts formation and up-regulation of the fibrosis-related proteins TGF-β1, connective tissue growth factor (CTGF), and fibronectin (FN) can be observed in Pkd1(flox/flox); Ksp-Cre mice. In an embryonic kidneys-cyst model, TGF-β1, FN and collagen type I were highly expressed. Western blotting revealed the obviously up-regulation of TGF-β1, CTGF, FN and collagen type I expression following forskolin treatment in MDCK cells. Selective PKA inhibition with H89 may partially reversed the above effects. Pretreatment with the TGF-β RI kinase inhibitor VI SB431542 suppressed the increased expression of CTGF, FN and collagen type I caused by forskolin. Our data also indicate that forskolin inhibited TGF-β-induced ERK1/2 phosphorylation and FN up-regulation. ERK inhibition useing PD98059 significantly inhibited the expression of CTGF, FN and collagen type I caused by TGF-β1.
CONCLUSIONS:
The cAMP-PKA signaling pathway can directly promote the production of TGF-β1 and/or TGF-β1-dependent fibrogenetic molecules in MDCK cells and embryonic kidney cysts, but when TGF-β1 and its downstream pathways were highly expressed in MDCK cells, cAMP-PKA had a significantly negative effect on TGF-β1 induced p-ERK1/2 and FN expression.
© 2015 S. Karger AG, Basel.
BACKGROUND:
Hyperglycemia is the most important risk factor in the progression of renal fibrosis in diabetic kidney. Based on previous studies, β-casomorphin-7 may exert anti-fibrotic activities in diabetic rats. However, the role of β-casomorphin-7 in the pathogenesis of renal tubulointerstitial fibrosis remains unclear. Thus, this study was designed to investigate the protective effect of β-casomorphin-7 on epithelial-mesenchymal transition (EMT) of NRK-52E cells treated under hyperglycemic condition and to explore the possible mechanism.
RESEARCH DESIGN AND METHODS:
NRK-52E cells were cultured in high glucose (30 mM) for 3 days. Different concentrations of β-casomorphin-7, naloxone (antagonist of opioid receptor) and losartan (antagonist of angiotensin II type I receptor) were added in the culture. Expression of α-smooth muscle actin (α-SMA), E-cadherin, vimentin and cytokeratin19 mRNA were determined by real-time PCR. Protein levels of E-cadherin and α-SMA were analyzed by Western blotting. The concentrations of angiotensin (Ang) II and transforming growth factor β1 (TGF-β1) in the culture medium were determined.
RESULTS:
High glucose-induced up-regulation of vimentin mRNA and α-SMA mRNA and protein were significantly inhibited by β-casomorphin-7. On the contrary, high glucose-induced down-regulation of cytokeratin19 mRNA and E-cad mRNA and protein was significantly reversed by β-casomorphin-7. β-casomorphin-7 significantly alleviate high glucose induced increase of AngII and TGF-β1 in the culture. Moreover, losartan significantly attenuated the expression of TGF-β1 and EMT of NRK-52E cells treated under hyperglycemic condition. But naloxone did not affect the EMT of NRK-52E cells treated by high glucose and β-casomorphin-7.
CONCLUSION:
We demonstrate that β-casomorphin-7 has the potential to inhibit high glucose-induced renal proximal tubular EMT partly by modulating AngII-TGF-β1 pathway, but not by opioid receptor.
Copyright © 2015 Elsevier Inc. All rights reserved.
BACKGROUND:
Transforming growth factor beta 1 (TGFβ1) can induce epithelial-mesenchymal transition (EMT) in various human cancers, but the complex mechanisms underlying this have not been fully elucidated. Inhibitor of DNA binding 1 (Id-1) has been identified as a novel marker of ovarian cancer progression. This study aims to investigate the role of Id-1 in TGFβ1-induced EMT in human ovarian cancer cells.
METHODS:
Ovarian cancer cells expressing or not expressing Id-1 were incubated with TGFβ1. Changes in the EMT markers E-cadherin, vimentin, N-cadherin, Id-1, and miR-29b were detected using western blotting and qPCR analyses. Wound healing, transwell migration, and invasion assays were performed in cells where Id-1 was either knocked down or overexpressed. The effects of transfecting miR-29b mimics and inhibitors on Id-1 mRNA and protein expression were assessed. The interaction between miR-29b and Id-1 was confirmed using a luciferase reporter assay.
RESULTS:
Id-1 expression was increased and miR-29b expression was repressed in TGFβ1-responsive ovarian cancer cells. Id-1 overexpression increases and Id-1 knockdown decreases cell migration and invasion capacities. Id-1 silencing leads to a partial blocking of TGFβ1-induced EMT. miR-29b negatively regulates Id-1 expression. Direct binding of miR-29b to the 3'UTR region of Id-1 was confirmed using a luciferase reporter assay.
CONCLUSION:
Id-1, a protein repressed by miR-29b, facilitates TGFβ1-induced EMT in human ovarian cancer cells and represents a promising therapeutic target for treating ovarian cancer.
© 2014 S. Karger AG, Basel.
BACKGROUND:
Genetically modified cells have been shown to be one of the most effective tumor vaccine strategies. However, in many cases, such as in melanoma, induction of a potent immune responses against the disease still remains a major challenge. Thus, novel strategies to reinforce tumor vaccine efficacy are needed. Using microRNA (miR) and Zinc-finger E-box binding homeobox (ZEB) have received much attention for potentially regulating tumor progression. To elicit a potent antitumor efficacy against melanoma, we used tumor vaccine in combination with miR200c overexpression or ZEB1 knockdown to assess the efficacy of treatment of murine melanoma.
METHODS:
B16F10 cell vaccine expressing interleukin 21 (IL-21) in the glycosylpho- sphatidylinositol (GPI)-anchored form (B16F10/GPI-IL-21) were developed. The vaccine was immunized into mice challenged by B16F10 cells or B16F10 cells stably transduced with lentiviral-miR200c (B16F10/miR200c) or transfected with the ZEB1-shRNA recombinant (B16F10/shZEB1) or the B16F10/GPI-IL-21 vaccine. The immune responses, tumorigenicity and lung metastasis in mice were evaluated, respectively.
RESULTS:
The vaccination with B16F10/GPI-IL-21 markedly increased the serum cytokine levels of IFN-γ, TNF-α, IL-4 and decreased TGF-β level as well as augmented the cytotoxicity of splenocytes in immunized mice compared with control mice. In addition, the tumor vaccine B16F10/GPI-IL-21 significantly inhibited the tumor growth and reduced counts of lung metastases in mice challenged by B16F10/GPI-IL-21, B16F10/shZEB1 and B16F10/miR200c respectively compared with the control mice challenged by B16F10 cells. The efficacy mechanisms may involve in reinforcing immune responses, increasing expression of miR200c, E-cadherin and SMAD-7 and decreasing expression of TGF-β, ZEB1, Vimentin and N-cadherin in tumor tissues from the immunized mice.
CONCLUSIONS:
These results indicate that the tumor vaccine B16F10/GPI-IL-21 in combination with miR200c overexpression or ZEB1 knockdown effectively inhibited melanoma growth and metastasis a murine model. Such a strategy may, therefore, be used for the clinical trials.