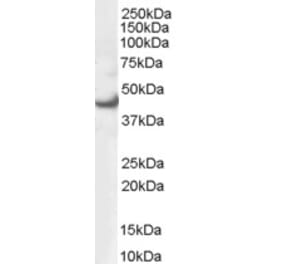
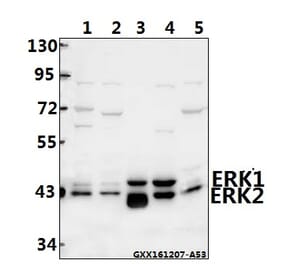
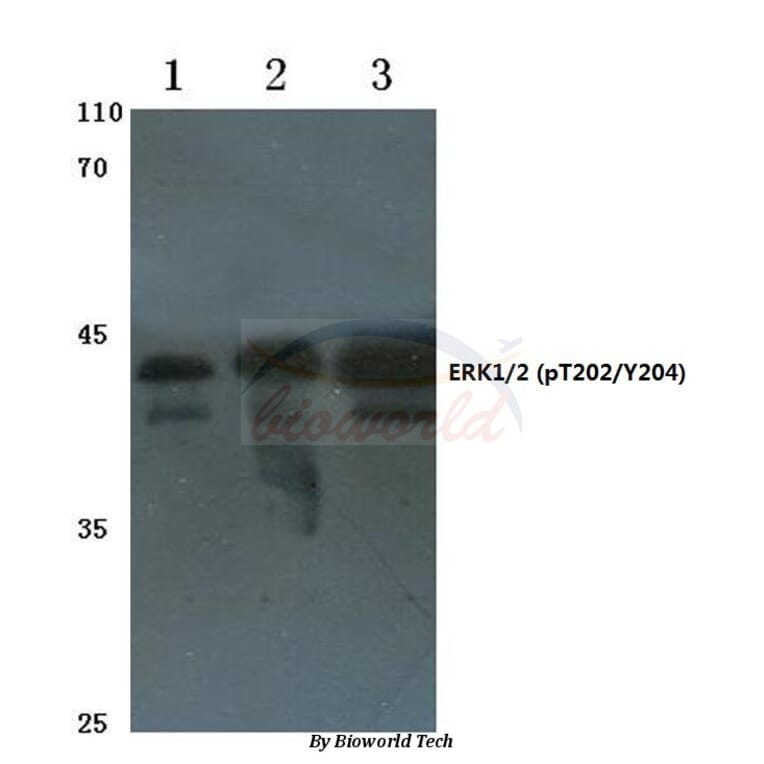
Unconjugated
Lipoxin A4 (LXA4), an endogenous arachidonic acid metabolite, was previously considered an anti-inflammatory lipid mediator. But it also has the potential to inhibit cancer progression. To explore the therapeutic effect of LXA4 in pancreatic cancer, we used Panc-1 cells to investigate the mechanism by which LXA4 can attenuate pancreatic cancer cell invasion. Our data showed that LXA4 significantly inhibited both cell invasion and the expression of matrix metalloproteinase- (MMP-) 9 and MMP-2. Further experiments implied that LXA4 decreased the levels of intracellular reactive oxygen species (ROS) and the activity of the extracellular signal regulated kinases (ERK) pathway to achieve similar outcome to ROS scavenger N-acetyl-L-cysteine (NAC). However, a decreased level of intracellular ROS was not observed in cells treated with the specific ERK pathway inhibitor FR180204. The blocking of either intracellular ROS or ERK pathway caused the downregulation of MMP-9 and MMP-2 expression. Furthermore, tests revealed that LXA4 inhibited MMP-9 and MMP-2 at the mRNA, protein, and functional levels. Finally, LXA4 dramatically limited the invasion of CoCl2-mimic hypoxic cells and abrogated intracellular ROS levels, ERK activity, and MMPs expression. These results suggest that LXA4 attenuates cell invasion in pancreatic cancer by suppressing the ROS/ERK/MMPs pathway, which may be beneficial for preventing the invasion of pancreatic cancer.
IL-6, a pleiotropic cytokine, has been investigated for its role in regulating autophagy. Yet, its mechanism of action remains unclear. Here, we show that IL-6 exerted anti-autophagic effects on U937 cells through the STAT3 signaling pathway in vitro. The addition of IL-6 to starved U937 cells significantly activated the phosphorylation level of STAT3 (p-STAT3) at Tyr705 and reduced the protein levels of microtubule-associated protein 1 light chain 3 of type II (LC3-II) and Beclin 1. By immunoblotting, we also observed a positive correlation between the p-STAT3 level and Bcl-2 level. Furthermore, treatment with a STAT3 inhibitor, LLL12, or overexpression of a mutant form, STAT3Y705F, reversed the inhibitory effect of IL-6 on autophagy. Knockdown of Beclin 1 or Atg14 by siRNA and over-expression of Beclin 1 indicated the involvement of class III PI3K complex in IL-6-mediated inhibition of autophagy. Taken together, these data indicate that IL-6 inhibits starvation-induced autophagy and that p-STAT3 mediates the signal transduction from IL-6 to downstream proteins including Bcl-2 and Beclin1.
Phloretin (Ph) existing in apples, pears and various vegetables is known to have antitumor activities in several cancer cell lines. However, little is known about its effect on human lung cancer cells. The aim of the present study was to see whether Ph could induce apoptosis of non-small cell lung cancer (NSCLC) cells, and explore the possible underlying mechanism of action. We found that Ph markedly induced cell apoptosis of NSCLC cell line A549, and inhibited the migration of A549 cells in a dose-dependent manner. The expression level of BAX, cleaved caspase-3 and -9, and degraded form of PARP was increased and Bcl-2 was decreased after Ph treatment. In addition, the phosphorylation of P38 MAPK, ERK1/2 and JNK1/2 was increased in a dose‑dependent manner in parallel with Ph treatment. Inhibition of P38 MAPK and JNK1/2 by specific inhibitors significantly abolished the Ph-induced activation of the caspase-3 and -9. In vivo tumor-suppression assay further indicated that Ph (20 mg/kg) displayed a more significant inhibitory effect on A549 xenografts in tumor growth. All these findings indicate that Ph is able to inhibit NSCLC A549 cell growth by inducing apoptosis through P38 MAPK and JNK1/2 pathways, and therefore may prove to be an adjuvant to the treatment of NSCLC.
Previous studies revealed that thymus is a targeted immune organ in malnutrition, and high-boron stress is harmful for immune organs. African ostrich is the living fossil of ancient birds and the food animals in modern life. There is no report about the effect of boron intake on thymus of ostrich. The purpose of present study was to evaluate the effect of excessive boron stress on ostrich thymus and the potential role of TLR3/4 signals in this process. Histological analysis demonstrated that long-term boron stress (640 mg/L for 90 days) did not disrupt ostrich thymic structure during postnatal development. However, the numbers of apoptotic cells showed an increased tendency, and the expression of autophagy and proliferation markers increased significantly in ostrich thymus after boron treatment. Next, we examined the expression of TLR3 and TLR4 with their downstream molecular in thymus under boron stress. Since ostrich genome was not available when we started the research, we first cloned ostrich TLR3 TLR4 cDNA from thymus. Ostrich TLR4 was close to white-throated Tinamou. Whole avian TLR4 codons were under purify selection during evolution, whereas 80 codons were under positive selection. TLR3 and TLR4 were expressed in ostrich thymus and bursa of fabricius as was revealed by quantitative real-time PCR (qRT-PCR). TLR4 expression increased with age but significantly decreased after boron treatment, whereas TLR3 expression showed the similar tendency. Their downstream molecular factors (IRF1, JNK, ERK, p38, IL-6 and IFN) did not change significantly in thymus, except that p100 was significantly increased under boron stress when analyzed by qRT-PCR or western blot. Taken together, these results suggest that ostrich thymus developed resistance against long-term excessive boron stress, possibly by accelerating intrathymic cell death and proliferation, which may bypass the TLR3/4 pathway. In addition, attenuated TLRs activity may explain the reduced inflammatory response to pathogens under boron stress.
Amplified in breast cancer 1 (AIB1) is a member of p160 steroid receptor coactivator (SRC) family that mediates the transcriptional activities of nuclear receptors and other transcription factors. It acts as a major oncogene in diverse cancers, whereas biological function of AIB1 in gastric cancer remains largely unclear. This study was designed to explore the role of AIB1 in gastric tumorigenesis and its potential as a useful prognostic marker and therapeutic target in this cancer. Our data demonstrated that AIB1 was significantly up-regulated in gastric cancer tissues as compared with control subjects. Moreover, AIB1 amplification was found in 47 of 133 (35.3%) gastric cancer cases, but not in control subjects. AIB1 amplification was positively associated with its protein expression, and was significantly correlated with poor patient survival. AIB1 knockdown in gastric cancer cells dramatically inhibited cell proliferation, invasiveness and tumorigenic potential in nude mice, and induced cell cycle arrest and apoptosis. Mechanically, AIB1 promotes gastric cancer cell proliferation, survival and invasiveness through modulating major signaling pathways such as ErbB and Wnt/β-catenin pathways. Collectively, these findings suggest that AIB1 plays an important role in the pathogenesis of gastric cancer and represents a potential prognostic marker and therapeutic target for this cancer.
Toll-like receptors (TLRs) recognize microbial pathogens and trigger immune response, but their regulation by neuropeptide-vasoactive intestinal peptide (VIP) in weaned piglets infected by enterotoxigenic Escherichia coli (ETEC) K88 remains unexplored. Therefore, the study was conducted to investigate its role using a model of early weaned piglets infected by ETEC K88. Male Duroc × Landrace × Yorkshire piglets (n = 24) were randomly divided into control, ETEC K88, VIP, and ETEC K88+VIP groups. On the first three days, ETEC K88 and ETEC K88+VIP groups were orally administrated with ETEC K88, other two groups were given sterile medium. Then each piglet from VIP and ETEC K88+VIP group received 10 nmol VIP intraperitoneally (i.p.) once daily, on day four and six. On the seventh day, the piglets were sacrificed. The results indicated that administration of VIP improved the growth performance, reduced diarrhea incidence of ETEC K88 challenged pigs, and mitigated the histopathological changes of intestine. Serum levels of IL-2, IL-6, IL-12p40, IFN-γ and TNF-α in the ETEC K88+ VIP group were significantly reduced compared with those in the ETEC group. VIP significantly increased IL-4, IL-10, TGF-β and S-IgA production compared with the ETEC K88 group. Besides, VIP could inhibit the expression of TLR2, TLR4, MyD88, NF-κB p65 and the phosphorylation of IκB-α, p-ERK, p-JNK, and p-38 induced by ETEC K88. Moreover, VIP could upregulate the expression of occludin in the ileum mucosa compared with the ETEC K88 group. Colon and caecum content bacterial richness and diversity were lower for pigs in the ETEC group than the unchallenged groups. These results demonstrate that VIP is beneficial for the maturation of the intestinal mucosal immune system and elicited local immunomodulatory activities. The TLR2/4-MyD88 mediated NF-κB and MAPK signaling pathway may be critical to the mechanism underlying the modulatory effect of VIP on intestinal mucosal immune function and bacterial community.
Emerging evidence indicate that mesenchymal stem cells (MSCs) affect tumor progression by reshaping the tumor microenvironment. Neutrophils are essential component of the tumor microenvironment and are critically involved in cancer progression. Whether the phenotype and function of neutrophils is influenced by MSCs is not well understood. Herein, we investigated the interaction between neutrophils and gastric cancer-derived MSCs (GC-MSCs) and explored the biological role of this interaction. We found that GC-MSCs induced the chemotaxis of neutrophils and protected them from spontaneous apoptosis. Neutrophils were activated by the conditioned medium from GC-MSCs with increased expression of IL-8, TNFα, CCL2, and oncostatin M (OSM). GC-MSCs-primed neutrophils augmented the migration of gastric cancer cells in a cell contact-dependent manner but had minimal effect on gastric cancer cell proliferation. In addition, GC-MSCs-primed neutrophils prompted endothelial cells to form tube-like structure in vitro. We demonstrated that GC-MSCs stimulated the activation of STAT3 and ERK1/2 pathways in neutrophils, which was essential for the functions of activated neutrophils. We further revealed that GC-MSCs-derived IL-6 was responsible for the protection and activation of neutrophils. In turn, GC-MSCs-primed neutrophils induced the differentiation of normal MSCs into cancer-associated fibroblasts (CAFs). Collectively, our results suggest that GC-MSCs regulate the chemotaxis, survival, activation, and function of neutrophils in gastric cancer via an IL-6-STAT3-ERK1/2 signaling cascade. The reciprocal interaction between GC-MSCs and neutrophils presents a novel mechanism for the role of MSCs in remodeling cancer niche and provides a potential target for gastric cancer therapy.
Insulin-like growth factor (IGF) signaling is involved in oral squamous cell carcinoma (OSCC), but IGF-1 receptor (IGF-1R)-mediated intricate regulatory networks among molecular interactions and signalling path ways in OSCC remain unclear. Here, we found that overexpression of IGF-1R and insulin receptor substrate-2 (IRS-2) was negatively associated with histological differentiation. IGF signaling stimulated OSCC cell growth. Conversely, overexpression of let-7b inhibited proliferation and colony formation and triggered S/G2 cell cycle arrest by targeting IGF-1R and IRS-2 through the Akt pathway. Also, the inverse relationship between expression of let-7b and IGF-1R/IRS-2 was confirmed in OSCC tumor xenografts and clinical specimens. Furthermore, by activating ERK1/2, IGF-1R transcriptionally upregulated IRS-2. Our results indicate that let-7b/IGF-1R-mediated crosstalk between IRS-2/Akt and MAPK is involved in OSCC and is a potential therapeutic target for therapy.
TNF-α is known to induce osteoblasts apoptosis, whereas mechanical stimulation has been shown to enhance osteoblast survival. In the present study, we found that mechanical stimulation in the form of fluid shear stress (FSS) suppresses TNF-α induced apoptosis in MC3T3-E1 cells. Extracellular signal-regulated kinase 5 (ERK5) is a member of the mitogen-activated protein kinase (MAPK) family that has been implicated in cell survival. We also demonstrated that FSS imposed by flow chamber in vitro leads to a markedly activation of ERK5, which was shown to be protective against TNF-α-induced apoptosis, whereas the transfection of siRNA against ERK5 (ERK5-siRNA) reversed the FSS-medicated anti-apoptotic effects. An initial FSS-mediated activation of ERK5 that phosphorylates AKT to increase its activity, and a following forkhead box O 3a (FoxO3a) was phosphorylated by activated AKT. Phosphorylated FoxO3a is sequestered in the cytoplasm, and prevents it from translocating to nucleus where it can increase the expression of FasL and Bim. The inhibition of AKT-FoxO3a signalings by a PI3K (PI3-kinase)/AKT inhibitor (LY294002) or the transfection of ERK5-siRNA led to the nuclear translocation of non-phosphorylated FoxO3a, and increased the protein expression of FasL and Bim. In addition, the activation of caspase-3 by TNF-α was significantly inhibited by aforementioned FSS-medicated mechanisms. In brief, the activation of ERK5-AKT-FoxO3a signaling pathways by FSS resulted in a decreased expression of FasL and Bim and an inhibition of caspase-3 activation, which exerts a protective effect that prevents osteoblasts from apoptosis.
Cancer associated fibroblasts (CAFs) are crucial contributors to breast cancer development. Estrogen affects mammary stroma in both physiological and pathophysiological conditions. We show here that estrogen (G-protein coupled) receptor (GPER) could be detected by immunohistochemistry in stromal fibroblasts of primary breast cancers. The presence of GPER expression was further confirmed by immunofluorescence and quantitative PCR in CAFs isolated from primary breast cancers. Based on dynamic monitoring by real time cell analyzer (RTCA) system, 17-β-estradiol (E2) as well as GPER specific agonist G1 were observed to trigger transient cell index increasing within an hour in a dosage-dependent manner in breast CAFs. In addition, E2 and G1 stimulated intracellular calcium modulation and phosphorylation of extracellular signal-regulated kinase (ERK) 1/2 within seconds and minutes in CAFs, respectively. Moreover, E2 and G1 promoted cell proliferation of breast CAFs measured by RTCA monitoring, cell viability assay and cell cycle analysis, and this promotion could be blocked by a GPER-selective antagonist G15. Interestingly, dynamic RTCA monitoring indicated that E2 increased adhesion of resuspended cells, and microscopy confirmed that E2 stimulated cell spreading. Both the adhesion and spreading were proposed to be mediated by GPER, since G1 also stimulated these effects similar to E2, and G15 reduced them. Moreover, GPER was found to mediate migration that was increased by E2 and G1 but reduced by G15 in RTCA cell migration assay and transwell assay. Accordingly, GPER mediates not only rapid actions but also slow effects including adhesion/spreading, proliferation and migration in breast CAFs. Estrogen is likely to affect tumor associated stroma and contributes to mammary carcinoma development through CAFs.
The proliferation of cardiac fibroblasts (CFs) and excessive deposition of extracellular matrix (ECM) are the main pathological characteristics of cardiac fibrosis. In recent years, microRNAs (miRNAs) have been found to be a new kind of regulator in cardiac fibrosis. The purpose of this study was to investigate the role of microRNA-9 (miR-9) in the process of cardiac fibrosis and its mechanism. Treatment of cultured neonatal rat CFs with PDGF-BB or serum suppressed the expression of miR-9. Overexpression of miR-9 obviously inhibited neonatal rat CFs proliferation and collagen production as detected by MTT assays, qRT-PCR, and western blotting. The effects of miR-9 in CFs were abrogated by co-transfection with miR-9 inhibitors. Overexpression of miR-9 reduced the mRNA and protein levels of PDGFR-βand its downstream protein, extracellular signal-regulated kinase (ERK) 1/2. Silencing PDGFR-βby small interfering RNA mimicked the anti-fibrotic action of miR-9, whereas overexpression of PGDFR-β canceled the effect of miR-9 in cultured CFs. Dual-luciferase reporter assays showed that PDGFR-βwas a direct target of miR-9. Overexpression of miR-9 inhibited cardiac fibrosis by targeting PDGFR-β, indicating that miR-9 might play a role in the treatment of cardiac fibrosis.
The anti-cancer effects of dioscin have been widely reported. However, its effect on laryngeal cancer remains unknown. In the present paper, our results showed that dioscin markedly caused cell apoptosis and DNA damage, increased reactive oxygen species (ROS) level, induced S-phase arrest, and inhibited invasion of human laryngeal cancer HEp-2 and TU212 cells. Mechanism investigation showed that dioscin markedly up-regulated p53 level, and down-regulated cyclin-dependent kinase 2 (CDK2) and Cyclin A levels. In addition, dioscin significantly down-regulated the levels of p-ERK, Bcl-2, up-regulated the levels of p-JNK, p-p38, Bax, cleaved caspase-3/-9, and caused Cytochrome c release. Furthermore, U0126, an ERK1/2 inhibitor, markedly down-regulated Bcl-2 level, up-regulated the levels of Bax, cleaved caspase-3/9, and enhanced Cytochrome c release inducted by dioscin. While, SP600125 (one JNK inhibitor) and SB203580 (one p38 inhibitor) markedly up-regulated Bcl-2 level, down-regulated the levels of Bax, cleaved caspase-3/9, and obviously boosted Cytochrome c release induced by dioscin. Interestingly, dioscin also markedly down-regulated the levels of MMP2 and MMP9 associated with tumor invasion. Taken together, our study indicated that dioscin suppressed laryngeal cancer cells growth via inducting cell-cycle arrest, MAPK-mediated mitochondrial- derived apoptosis and inhibiting tumor invasion, which could be used as one potential candidate for the treatment of laryngeal cancer in the future.
The involvement of miR-335 in csolorectal cancer (CRC) development remains controversial. Here, we found that miR-335 was highly up-regulated in CRC specimens relative to normal mucosa, and high miR-335 expression level was markedly associated with the tumour size and differentiation of CRC. The overexpression of miR-335 in CRC cells facilitated cell proliferation in vitro and tumour growth in vivo. RASA1 was validated as a target of miR-335 that was downregulation in CRC. Forced expression of miR-335 silenced RASA1 and triggered Ras/ERK cascade in CRC. Together, miR-335-RASA1 contributes to cell growth in CRC, and elucidation of downstream pathway will provide new insights into the molecular mechanisms of CRC progression.
Endotoxin can stimulate inflammatory cytokine release from monocytes/macrophages and result in septic shock. Glycyrrhetinic acid (GA), the main bioactive component of licorice, possesses substantial anti-inflammatory activity. Here, we explored effect of 11-deoxy-18α-glycyrrhetinic acid-30-ethyl ester (DGAEE), a newly synthesized derivative of GA, on septic shock. DGAEE and its main metabolite 11-deoxy-18α-glycyrrhetinic acid (DGA) significantly alleviated septic shock as evidenced by improvements of survival rates, lung histopathological changes and wet/dry ratio in lipopolysaccharide (LPS)/D-galactosamine-stimulated mice, and decreased blood pressure in LPS/D-galactosamine-stimulated rats. The two compounds decreased serum levels of NO, TNF-α, IL-6, IL-1β, and increased the level of IL-10 more potently in mice. In LPS-stimulated RAW 264.7 cells, DGA but not DGAEE showed marked regulation of NO, TNF-α, IL-6 and IL-10 levels, suggesting that DGAEE display anti-shock effect by DGA rather than itself. Moreover, the neutralizing antibody against IL-10 markedly prohibited the inhibitory effect of DGA on the production of cytokines from RAW 264.7 cells, and AS101 (an inhibitor of IL-10 biosynthesis) almost completely reversed the anti-shock effect of DGA in mice. In addition, DGA did not affect activation of NF-κB-p65 and p38 MAPK as well as IκBα degradation, but moderately reduced activation of ERK and JNK, and markedly increased phosphorylation of GSK3β in LPS-stimulated RAW 264.7 cells. LY294002 (an inhibitor of GSK3β phosphorylation) and LiCl (an inhibitor of GSK3β activity) diminished and potentiated increase of IL-10 levels by DGA, respectively. In conclusion, DGAEE alleviates septic shock through DGA in an IL-10-dependent manner, and the mechanism is related to inactivation of GSK3β.
Norisoboldine (NOR), the main active constituent of Radix Linderae, was previously demonstrated to ameliorate collagen-induced arthritis in rats through regulating the imbalance of T cells in intestines, which implied its therapeutic potential in inflammatory bowel disease. Here, we investigated the effect of NOR on ulcerative colitis (UC) induced by dextran sulfate sodium (DSS) in mice. Results showed that NOR (20, 40mg/kg) markedly reduced the symptoms of colitis, the levels of IL-1β and TNF-α, and the activation of ERK, p38 MAPK and NF-κB-p65. NOR only slightly decreased the levels of IFN-γ and IL-17A in mouse colons, but it dramatically increased the level of IL-10 at both protein and mRNA grades. Consistently, NOR increased the number of CD4(+)CD25(+)Foxp3(+) Treg cells more obviously than it decreased that of CD4(+)IL-17(+) Th17 cells in mesenteric lymph nodes (MLNs) and colonic lamina proprias (LPs) of colitis mice, and promoted the expression of Foxp3 mRNA in colon tissues. It could facilitate the in vitro differentiation of Treg cells from naive T cells and promote the phosphorylations of Smad2/3 in colon tissues of colitis mice. On the other hand, NOR did not affect the expressions of homing receptors CCR9 and α4β7 in SPs, and homing ligands CCL25 and Madcam-1 in MLNs and colonic LPs, suggesting that the increase of Treg cells in colons by NOR was not due to gut homing. In conclusion, NOR can ameliorate DSS-induced UC in mice, and the mechanisms involve reduction of pro-inflammatory cytokines and selective induction of Treg cells in colons.
Fluid shear stress (FSS) is a potent mechanical stimulus and prevents cells from TNF-a-induced apoptosis. Recently, Extracellular-signal-regulated kinase 5 (ERK5) has been found to be involved in regulation of cell survival. However, little is known about the role of ERK5 signaling pathway in FSS-mediated anti-apoptotic effects in osteoblast. In this study, we show that FSS blocks TNF-a-induced apoptosis of MC3T3-E1 cells via ERK5 signaling pathway. We found that physiological FSS for 1 h significantly decreased TNF-α-induced MC3T3-E1 cells apoptosis. After inhibition of ERK5 activity by XMD8-92, a highly-selective inhibitor of ERK5 activity, the ability of FSS to inhibit TNF-α induced apoptosis was significantly decreased. Analysis of anti-apoptotic mechanisms indicated that exposure of MC3T3-E1 cells to FSS for 1 h increased phosphorylation of Bad and inhibited caspase-3 activity. After treatment with XMD8-92, phosphorylation of Bad by FSS was significantly blocked, but caspase-3 activity was increased. In summary, these findings indicated that FSS inhibits TNF-α-mediated signaling events in osteoblast by a mechanism dependent on activation of ERK5, and Bad is a crucial downstream target for ERK5. Those results implied that ERK5 signaling pathway play a crucial role in FSS-mediated anti-apoptotic effect in osteoblast. Thus, ERK5 signaling pathway may be a new drug treatment target of osteoporosis and related bone-wasting diseases.
This study investigated anti-inflammatory effects and possible mechanisms of Chikusetsusaponin IVa (Chi IVa), one of the main bioactive components in saponins from Panacis japonica (SPJ), which is used in traditional Tujia and Hmong Chinese medicine. To this end, changes in the inflammatory profiles of lipopolysacchride (LPS)-stimulated phrobol 12-myristate 13-acetate(PMA)-differented THP-1 macrophages were evaluated following Chi IVa treatment. The results showed that Chi IVa markedly decreased the expression of inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), interleukin-1 beta (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) at both the mRNA and protein level, which proved to be dose-dependent. Further studies revealed that Chi IVa strongly suppressed NF-κB activation and downregulated the phosphorylation of ERK, p38, and JNK. Our present study demonstrates that Chi IVa suppresses the production of iNOS, COX-2, IL-1β, IL-6, and TNF-α in LPS-stimulated THP-1 cells likely by inhibiting NF-κB activation and ERK, JNK, and p38 signal pathway phosphorylation.
Ruscogenin, a natural steroidal sapogenin, presents in both food and medicinal plants. It has been found to exert significant anti-inflammatory activities. Considering that activation of neutrophil is a key feature of inflammatory diseases, this study was performed to investigate the inhibitory effect of ruscogenin and its underlying mechanisms responsible for neutrophil activation. Ruscogenin displayed potent antioxidative effects against Formyl-Met-Leu-Phe (FMLP)-induced extra- and intracellular superoxide generation in mouse bone marrow neutrophils, with IC50 values of 1.07±0.32 μM and 1.77±0.46 μM, respectively. Phorbol myristate acetate (PMA)-elicited extra- and intracellular superoxide generation were also suppressed by ruscogenin, with IC50 values of 1.56±0.46 μM and 1.29±0.49 μM, respectively. However, ruscogenin showed weak inhibition in NaF-induced response. Inhibition of superoxide generation was mediated neither by a superoxide-scavenging ability nor by a cytotoxic effect. Furthermore, ruscogenin inhibited the membrane translocation of p47phox and p67phox. It reduced FMLP-induced phosphorylation of cytosolic phospholipase A2 (cPLA2) and p21-activated kinase (PAK). The cellular cyclic adenosine monophosphate (cAMP) levels and protein kinase A (PKA) expression were increased by ruscogenin. Moreover, ruscogenin inhibited phosphorylation of protein kinase B (Akt), p38 mitogen-activated protein kinase (p38MAPK), extracellular signal-regulated kinase 1 and 2 (ERK1/2), and c-Jun N-terminal kinase (JNK). In addition, the inhibitory effects of ruscogenin on superoxide production and the phosphorylation of Akt, p38MAPK, and ERK1/2 were reversed by PKA inhibitor (H89), suggesting a PKA-dependent mechanism. In summary, our data suggest that ruscogenin inhibits activation of neutrophil through cPLA2, PAK, Akt, MAPKs, cAMP, and PKA signaling pathways. Increased PKA activity is associated with suppression of the phosphorylation of Akt, p38MAPK, and ERK1/2 pathways.
Adrenomedullin (ADM), a multifunctional regulatory peptide, is potentially induced by hypoxia in physiological and pathological tissues, including many types of malignant tumors. Recent research has demonstrated that ADM expression is highly associated with the prognosis and disease severity of human osteosarcoma. However, the effect of ADM on the apoptosis of osteosarcoma cells and its possible mechanism remain to be elucidated. In the present study, we observed that mRNA and protein levels of ADM were increased in human osteosarcoma SOSP-F5M2 cells under a hypoxic microenvironment induced by cobalt chloride (CoCl2) in a time-dependent manner. Treatment with ADM significantly blunted hypoxic-induced apoptosis, evaluated by Hoechst 33342 staining and Annexin V-FITC/PI labeling. The expression of B-cell lymphoma-2 (Bcl-2) was increased by administration of ADM; meanwhile, this effect was reversed by exogenously adding U0126, a selective inhibitor of MEK or ADM22-52 (ADM-specific receptor antagonist). These results demonstrated that ADM acted as a survival factor to inhibit hypoxic-induced apoptosis via interacting with its receptors CRLR-RAMP (2,3) in osteosarcoma cells. The anti-apoptotic function of ADM was found to be mediated by upregulation of the expression of Bcl-2 partially through activation of the MEK/ERK1/2 signaling pathway. Therefore, targeting of the ADM/ADM acceptors/ERK1/2/Bcl-2 pathway may provide a potential strategy through which to induce the apoptosis of osteosarcoma cells.
The aim of this study was to explore the intracellular mechanisms underlying the cardiovascular toxicity of air particulate matter (PM) with an aerodynamic diameter of less than 2.5 µm (PM2.5) in a human umbilical vein cell line, EA.hy926. We found that PM2.5 exposure triggered reactive oxygen species (ROS) generation, resulting in a significant decrease in cell viability. Data from Western blots showed that PM2.5 induced phosphorylation of Jun N-terminal kinase (JNK), extracellular signal regulatory kinase (ERK), p38 mitogen-activated protein kinase (MAPK) and protein kinase B (AKT), and activation of nuclear factor kappa B (NF-κB). We further observed a significant increase in expressions of intercellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1) in a time- and dose-dependent manner. Moreover, the adhesion of monocytic THP-1 cells to EA.hy926 cells was greatly enhanced in the presence of PM2.5 . However, N-acetylcysteine (NAC), a scavenger of ROS, prevented the increase of ROS generation, attenuated the phosphorylation of the above kinases, and decreased the NF-κB activation as well as the expression of ICAM-1 and VCAM-1. Furthermore, ERK inhibitor (U0126), AKT inhibitor (LY294002) and NF-κB inhibitor (BAY11-7082) significantly down-regulated PM2.5 -induced ICAM-1 and VCAM-1 expression as well as adhesion of THP-1 cells, but not JNK inhibitor (SP600125) and p38 MAPK inhibitor (SB203580), indicating that ERK/AKT/NF-κB is involved in the signaling pathway that leads to PM2.5 -induced ICAM-1 and VCAM-1 expression. These findings suggest PM2.5 -induced ROS may function as signaling molecules triggering ICAM-1 and VCAM-1 expressions through activating the ERK/AKT/NF-κB-dependent pathway, and further promoting monocyte adhesion to endothelial cells.
Vitexin is a major bioactive flavonoid compound derived from the dried leaf of hawthorn (Crataegus pinnatifida), a widely used conventional folk medicine in China. Recent studies have shown that vitexin presents neuroprotective effects in vitro. Whether this protective effect applies to the cerebral ischemia/reperfusion (I/R) injury remains elusive. In the present study, we examined the potential neuroprotective effect of vitexin against cerebral I/R injury and underlying mechanisms. A focal cerebral I/R model in male Kunming mice was induced by middle cerebral artery occlusion (MCAO) for 2 h followed by reperfusion for 22 h. The neurological function and infarct volume were assessed by using Long's five-point scale system and triphenyl-tetrazolium chloride (TTC) staining technique, respectively. Neuronal damage was evaluated by histological staining. Extracellular signal-regulated kinases 1/2 (ERK1/2), c-Jun N-terminal kinases (JNK) and p38 phosphorylation, and apoptosis were measured via Western blot at 24 h after reperfusion. As a result, systemic vitexin treatment significantly reduced neurological deficit, cerebral infarct volume and neuronal damage when compared with the I/R group. Western blot analyses revealed that vitexin markedly upregulated p-ERK1/2 and downregulated p-JNK and p-p38. Meanwhile, vitexin increased Bcl-2 expression and suppressed the overexpression of Bax in the I/R injury mice. In conclusion, the results indicate that vitexin protects brain against cerebral I/R injury, and this effect may be regulated by mitogen-activated protein kinase (MAPK) and apoptosis signaling pathways.
Inflammation in the brain, characterized by the activation of microglia, is believed to participate in the pathogenesis of Parkinson's disease. Biochanin A, an O-methylated isoflavone, is a natural organic compound and is classified as a phytoestrogen. In this study, using murine BV2 microglial cells, we investigated the anti-inflammatory effects of biochanin A and the possible mechanisms involved. BV2 microglial cells were treated with lipopolysaccharide (LPS) to induce pro-inflammatory responses and the cells were then treated with biochanin A. Cell viability was examined by MTT assay. The production of nitric oxide (NO) was examined using Griess reagent and intracellular reactive oxygen species (ROS production) was measured by DCFH-DA assay. The mRNA expression of interleukin-1β (IL-1β), inducible nitric oxide synthase (iNOS) and tumor necrosis factor-α (TNF-α) was examined by RT-PCR. The expression of p-ERK, p-JNK, p-p38 and iNOS was measured by western blot analysis. In addition, the protein and mRNA and phosphorylation levels of pro-inflammatory cytokines were determined by western blot analysis and RT-PCR, respectively. The results revealed that biochanin A attenuated LPS-induced microglial activation and the production of TNF‑α, IL-1β, nitric oxide and reactive oxygen species in a dose-dependent manner. Biochanin A significantly decreased the LPS-induced mRNA expression of TNF-α and IL-1β, and inhibited iNOS mRNA and protein expression. Furthermore, biochanin A significantly inhibited the LPS-induced phosphorylation of c-Jun NH2-terminal kinase (JNK), extracellular signal-regulated kinase (ERK) and p38. These findings suggest that the inhibitory effects of biochanin A on LPS-induced proinflammatory responses may be associated with the inhibition of mitogen-activated protein kinase (MAPK) signaling pathways in BV2 microglial cells.
Obovatol, a compound isolated from the bark cortex of Magnolia officinalis (cortex Magnoliae officinalis; M. officinalis), has been studied for use in the treatment of solid cancers. However, the mechanisms of action and the effects of obovatol against acute myeloid leukemia (AML) remain unclear and require further investigation. Therefore, this study was conducted using a human AML cell line (MM6). Obovatol increased pro-apoptotic (Bax) and decreased anti-apoptotic (Bcl-2) protein expression, resulting in caspase-3 and caspase-9 activation measured by caspase-Glo 3/7 assay. Furthermore, obovatol activated the mitogen-activated protein kinase (MAPK) signaling pathway [c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK) and p38] and inhibited the activation of the nuclear factor-κB (NF-κB) signaling pathway analyzed by western blot analysis. Taken together, these findings provide evidence that obovatol inhibits cell proliferation in AML and induces apoptosis through the activation of the MAPK pathway in addition to the intrinsic apoptotic pathway. In addition, obovatol suppressed the expression of mixed-lineage leukemia (MLL) target genes by inhibiting the activation of the NF-κB pathway. Therefore, these results suggest that obovatol may have potential for use in the treatment of leukemia.
Diosgenin possesses anti-inflammatory and anticancer properties. Activated neutrophils produce high concentrations of the superoxide anion which is involved in the pathophysiology of inflammation-related diseases and cancer. In the present study, the inhibitory effect and possible mechanisms of diosgenin on superoxide generation were investigated in mouse bone marrow neutrophils. Diosgenin potently and concentration-dependently inhibited the extracellular and intracellular superoxide anion generation in Formyl-Met-Leu-Phe (FMLP)- activated neutrophils, with IC50 values of 0.50 ± 0.08 μM and 0.66 ± 0.13 μM, respectively. Such inhibition was not mediated by scavenging the superoxide anion or by a cytotoxic effect. Diosgenin inhibited the phosphorylation of p47phox and membrane translocation of p47phox and p67phox, and thus blocking the assembly of nicotinamide adenine dinucleotide phosphate oxidase. Moreover, cellular cyclic adenosine monophosphate (cAMP) levels and protein kinase A (PKA) expression were also effectively increased by diosgenin. It attenuated FMLP-induced increase of phosphorylation of cytosolic phospholipase A (cPLA2), p21-activated kinase (PAK), Akt, p38 mitogen-activated protein kinase (p38MAPK), extracellular signal-regulated kinase (ERK1/2), and c-Jun N-terminal kinase (JNK). Our data indicate that diosgenin exhibits inhibitory effects on superoxide anion production through the blockade of cAMP, PKA, cPLA2, PAK, Akt and MAPKs signaling pathways. The results may explain the clinical implications of diosgenin in the treatment of inflammation-related disorders.
Ubiquitin‑specific protease 22 (USP22) is a component of the transcription regulatory histone acetylation complex SAGA, which broadly regulates gene transcription and correlates with cancer progression, metastasis and prognosis. Autophagy is a cell pathway with dual functions that promotes cell survival or death. However, it is not known whether USP22 can regulate autophagy in pancreatic cancer. In the present study, we first identified that USP22 was overexpressed in a large number of pancreatic cancer patient samples, concomitant with the increased expression of LC3, a marker of autophagy. Statistical analysis revealed that the increase in USP22 and autophagy was positively correlated with poor prognosis of pancreatic cancer patients. Further investigation using a human pancreatic cancer cell (Panc‑1) identified that the overexpression of USP22 increased the processing of LC3 into the active form LC3‑II and the number of autophagosomes, thus leading to enhanced autophagy. Activation of ERK1/2 kinase rather than AKT1 by USP22 was found to be one of the mechanisms promoting LC3 processing. USP22‑induced autophagy was also found to enhance cell proliferation and resistance to starvation and chemotherapeutic drugs in Panc‑1 cells, therefore expressing an overall effect that promotes cell survival. Collectively, the present study demonstrated a new function of USP22 that induces autophagy, thus leading to the poor prognosis of pancreatic cancer.
Chronic exposure to fluoride can result in a variety of adverse effects in fish. Previously we indicated that high fluoride caused damage and apoptosis in the kidneys of the common carp, Cyprinus carpio. In this study, the effects of fluoride on the expression and localization of ERK and p-ERK proteins in the ERK signaling pathway were determined using Western blotting and immunohistochemical methods in the kidneys of carp exposed to 0, 40, 80, 120mg/L fluoride, respectively. Western blotting analysis found that compared with the controls, the levels of ERK1 and ERK2 proteins were relatively unchanged in fluoride-exposed fish, while p-ERK1 and p-ERK2 protein levels decreased significantly with the increased fluoride concentrations. The immunohistochemical analysis found the proteins of ERK and p-ERK were predominantly localized in the cytoplasm of epithelial cells in the renal tubules of C. carpio. Compared with the control group, the levels of ERK protein were relatively constant, yet the levels of p-ERK protein and p-ERK/ERK ratio were reduced with fluoride exposure dose. These findings indicate that the renal damage in carp exposed to fluoride is mediated via the ERK pathway. Fluoride exposure could inactivate ERK, inhibit the expression of p-ERK protein, and induce renal damage in C. carpio.
Melatonin, a major pineal secretory product, exerts a range of physiological and neuroprotective effects. However, the functional significance of melatonin in determining neural identity, and the mechanisms by which this may occur, is unknown. In this study, P19 cells were used as a model system and cell behavior was monitored. Our data show that melatonin plays an important role in determining cell fate during neural commitment and promoting the differentiation of pluripotent P19 cells (Oct4(+) Sox2(+) ) into neural stem cells (Oct4(-) Sox2(+) ). This promotion appears to coincide with the activation of the MT1 receptor and phosphorylation of extracellular-signal-regulated kinases 1/2 (ERK1/2). Furthermore, our results show that melatonin regulates neural fate specification of P19 cells through two distinct mechanisms: the promotion of nuclear localization of ERK1/2 and upregulation of Sox2 transcription, and suppression of Smad1-induced expression of mesodermal-specific genes, such as Bra.
This study was conducted to demonstrate myocardial protective effects and possible underlying mechanisms of vitexin on myocardial ischemia/reperfusion (I/R) injury in rats. Occluding the anterior descending artery for 30 min and restoring blood perfusion for 60 min in rat established a model of myocardial I/R. The elevation of the ST segment of Electrocardiograph (ECG) was observed. The infarct size of the rat heart was assessed by triphenyltetrazolium chloride staining (TTC). LDH, CK, SOD activities and MDA content were determined. An immunohistochemical analysis was applied to measure the expression of myocardial NF-κBp65 and TNF-α. ERK/phospho-ERKand c-Jun/phospho-c-Jun protein expression was examined via Western Blot. Vitexin significantly reduced the elevation of the ST segment of ECG and myocardial infarct size. LDH and CK activities and MDA content were attenuated in serum, while SOD activity was markedly enhanced. Vitexin significantly attenuated I/R-induced increases of myocardial NF-κB and TNF-α. Moreover, Western Blot analysis presented that vitexin markedly enhanced the expression of phospho-ERK and weakened the expression of phospho-c-Jun compared to I/R group. The significant protective effect against myocardial ischemical/reperfusion injury in rat, which is exhibited by vitexin, may be related to its antioxidative and anti-inflammatory effects by regulating inflammatory cytokines and the MAPK pathway.
The peptide amphiphile (PA) with a laminin epitope IKVAV (IKVAV-PA) can be trigged into three-dimensional nanostructures in vivo. Application of IKVAV-PA to the injured spinal cord resulted in significant functional improvement in rodents with remarkable axonal regeneration at the lesion site. Here we showed that injection of IKVAV-PA into the hippocampus of a transgenic (Tg) mice model of Alzheimer's disease (AD) significantly improved cognitive impairment, accompanied by an enhanced neurogenesis in the hippocampus. Further examination demonstrated that IKVAV-PA injections also significantly reduced the levels of soluble Aβ1-40, Aβ1-42, and amyloid-beta (Aβ) plaques in these brains. Our data suggest that IKVAV-PA may serve as a potential therapeutic intervention for the learning and memory losses in AD.
The RNA-binding protein Musashi2 (Msi2) has been identified as a master regulator within a variety of stem cell populations via the regulation of translational gene expression. A recent study has suggested that Msi2 is strongly expressed in leukemic cells of acute myeloid leukemia patients, and elevated Msi2 is associated with poor prognosis. However, the potential role of Msi2 in leukemogenesis is still not well understood. Here, we investigated the effect of Msi2 knockdown on the biological properties of leukemic cells. High expression of Msi2 was found in K562 and KG-1a leukemic cell lines, and low expression was observed in the U937 cell line. We transduced K562 cells with two independent adenoviral shRNA vectors targeting Msi2 and confirmed knockdown of Msi2 at the mRNA and protein levels. Msi2 silencing inhibited cell growth and caused cell cycle arrest by increasing the expression of p21 and decreasing the expression of cyclin D1 and cdk2. In addition, knockdown of Msi2 promoted cellular apoptosis via the upregulation of Bax and downregulation of Bcl-2 expression. Furthermore, Msi2 knockdown resulted in the inactivation of the ERK/MAPK and p38/MAPK pathways, but no remarkable change in p-AKT was observed. These data provide evidence that Msi2 plays an important role in leukemogenesis involving the MAPK signaling pathway, which indicates that Msi2 may be a novel target for leukemia treatment.
Fluid shear stress protects cells from TNF-α-induced apoptosis. Oscillatory fluid shear stress (OFSS) is generally perceived as physiologically relevant biophysical signal for bone cells. Here we identify several cellular mechanisms responsible for mediating the protective effects of OFSS against TNF-α-induced apoptosis in vitro. We found that exposure of MC3T3-E1 osteoblast-like cells to as little as 5 min of OFSS suppressed TNF-α-induced activation of caspase-3, cleavage of PARP and phosphorylation of histone. In contrast, H(2)O(2)-induced apoptosis was not inhibited by OFSS suggesting that OFSS might not be protecting cells from TNF-α-induced apoptosis via stimulation of global pro-survival signaling pathways. In support of this speculation, OFSS inhibition of TNF-α-induced apoptosis was unaffected by inhibitors of several pro-survival signaling pathways including pI3-kinase (LY294002), MAPK/ERK kinase (PD98059 or U0126), intracellular Ca2+ release (U73122), NO production (L-NAME), or protein synthesis (cycloheximide) that were applied to cells during exposure to OFSS and during TNF-α treatment. However, TNF-α-induced phosphorylation and degradation of IκBα was blocked by pre-exposure of cells to OFSS suggesting a more specific effect of OFSS on TNF-α signaling. We therefore focused on the mechanism of OFSS regulation of TNF-receptor 1 (TNFR1) signaling and found that OFSS (1) reduced the amount of receptor on the cell surface, (2) prevented the association of ubiquitinated RIP in TNFR1 complexes with TRADD and TRAF2, and (3) reduced TNF-α-induced IL-8 promoter activity in the nucleus. We conclude that the anti-apoptotic effect of OFSS is not mediated by activation of universal pro-survival signaling pathways. Rather, OFSS inhibits TNF-α-induced pro-apoptotic signaling which can be explained by the down-regulation of TNFR1 on the cell surface and blockade of TNFR1 downstream signaling by OFSS.
RATIONALE:
The mechanism involved in AD is complex, which has prompted to develop compounds that could simultaneously interact with several potential targets. Here, we report a new synthesized compound SCR-1693 which is designed to target both AChE and calcium channels that are potential for AD therapy.
OBJECTIVES:
We investigated the effects of SCR-1693 on AChE and calcium channels, the effects of neuroprotection and anti-amnesia in icv-Aβ25-35-injected mice, and the potential mechanisms.
METHODS:
AChE activity assay, intracellular Ca(2+) content and calcium currents measurement, and Aβ25-35-induced cellular death determine were performed for validation of designed targets and neuroprotection of SCR-1693. Mice were orally administrated with SCR-1693 once daily after an Aβ25-35 injection. The Morris water maze and Y-maze test, and hippocampal protein detection were conducted on days 5-10, day 11, and day 8. The pyramidal neuron number, hippocampal AChE activity, and synaptic transmission were measured on day 12.
RESULTS:
SCR-1693 acted as a selective, reversible, and noncompetitive inhibitor of AChE, and a nonselective voltage-gated calcium channel blocker. SCR-1693 also inhibited the increase of AChE activity in the mouse hippocampus. SCR-1693 was more effective than donepezil and memantine in preventing Aβ25-35-induced long-term and short-term memory impairment, maintaining the basal transmission of Schaffer collateral-CA1 synapses, and sustaining LTP in mouse hippocampus. SCR-1693 attenuated Aβ25-35-induced death of SH-SY5Y cell and the loss of hippocampal pyramidal neurons, and regulated Aβ25-35-induced signal cascade in neurons.
CONCLUSIONS:
All these findings indicated that SCR-1693, as a double-target-direction agent, is a considerable candidate for AD therapy.
BACKGROUND/AIMS:
8-Methoxypsoralen (8-MOP), a formerly considered photosensitizing agent, induces apoptosis when used alone. On this basis, the present study was designed to explore the effects and mechanisms of 8-MOP-induced apoptosis in human hepatocellular carcinoma HepG2 cells, independent of its photoactivation.
METHODS:
We analyzed the cell viability with MTT assay. Flow cytometry was used to examine the apoptosis rate, mitochondrial membrane potential (MMP) and reactive oxygen species (ROS) generation after specific staining. The expression and location of apoptosis-associated protein as well as the activation status of cell signaling pathway were determined by Western blot analysis.
RESULTS:
8-MOP significantly decreased cell viability and induced cell apoptosis through mitochondrial apoptotic pathway, as demonstrated by increased Bax/Bcl-2 ratio, collapsed MMP, and induced cytochrome c release (Cyt c) and apoptosis-inducing factor (AIF) transposition. ROS generation was significantly increased by 8-MOP and the eradication of ROS significantly abolished 8-MOP-induced apoptosis. In addition, the activation of ERK1/2 was drastically decreased by 8-MOP as ERK inhibitor PD98059, indicating a role of ERK1/2 signaling pathway in 8-MOP-induced cell apoptosis.
CONCLUSION:
8-MOP induces intrinsic apoptosis by increasing ROS generation and inhibiting ERK1/2 pathway in HepG2 cells. The findings are important in substantiating the anti-tumor role of 8-MOP in cancer therapy.
© 2015 S. Karger AG, Basel.
AIM:
Inflammation and oxidative stress are now recognized to be two important contributing factors to the development of atherosclerosis(AS). NADPH oxidase-4 (Nox4)-derived reactive oxygen species(ROS), NF-κB and MAPK play crucial roles in these processes. Luteolin, a flavone rich in many plants, can interrupt the molecular expression and inhibit the progression of inflammation and oxidative stress. The present study was designed to test whether luteolin inhibits TNF-α-induced inflammation and oxidative stress in human umbilical vein endothelial cells(HUVECs) and identify some of the mechanisms underlying these effects.
METHODS:
HUVECs were treated with luteolin in the presence/absence of TNF-α. The mechanism of luteolin against TNF-α-induced cell injury was evaluated using Western blotting, real-time RT-PCR and flow cytometry analyses.
RESULTS:
Luteolin suppressed the TNF-α-activated ROS generation, as well as the Nox4, p22phox, and ICAM-1 and VCAM-1 expression. Luteolin also enhanced the Bcl-2 and reduced caspase-3, -9 expression in the TNF-α-treated HUVECs. Finally, luteolin inhibited the TNF-α-induced transcriptional activity of NF-κB and p38 in addition to ERK1/2 phosphorylation. The inhibitors and siRNA of Nox4 and NF-κB not only reduced ROS generation, p38, ERK1/2 phosphorylation and the ICAM-1 and VCAM-1 expression, but also enhanced Bcl-2 expression. The inhibitor of p38 had the same effect on the expression of ICAM-1, VCAM-1 and Bcl-2, while the inhibitor of ERK1/2 increased the Bcl-2 expression rather than reducing the ICAM-1 and VCAM-1 expression.
CONCLUSIONS:
Luteolin attenuates TNF-α-induced oxidative stress and inflammation via its effects on the Nox4/ROS-NF-κB and MAPK pathways. These results suggest that luteolin may provide a beneficial effect in treating vascular diseases associated with oxidative stress and inflammation.
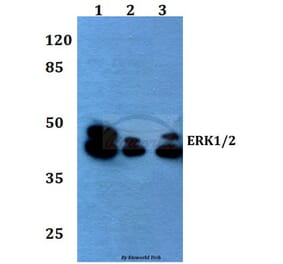
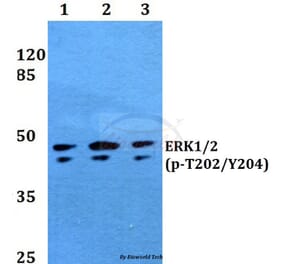
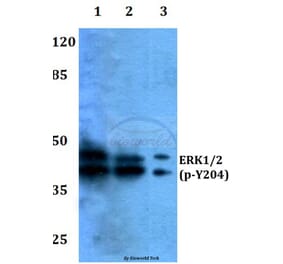
![Western Blot - Anti-ERK1 Antibody [ARC2591] (A306776) - Antibodies.com](https://cdn.antibodies.com/image/catalog/306/A306776_1.jpg?profile=product_alternative)