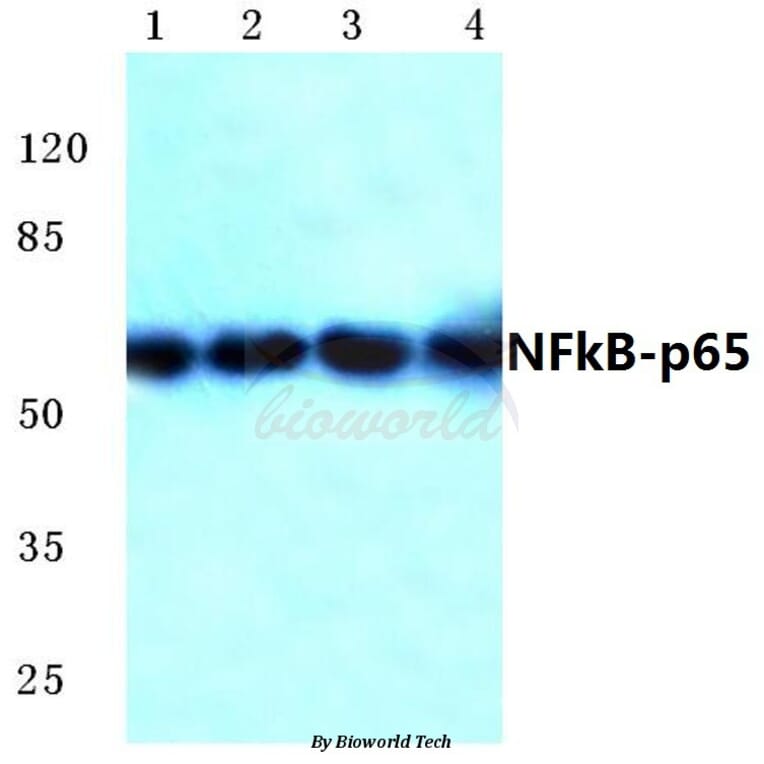
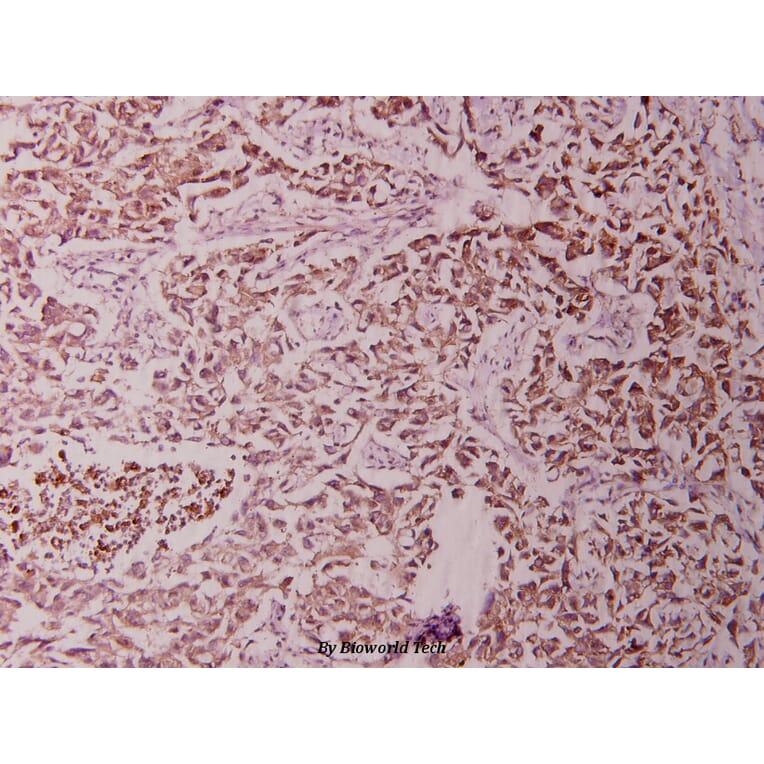
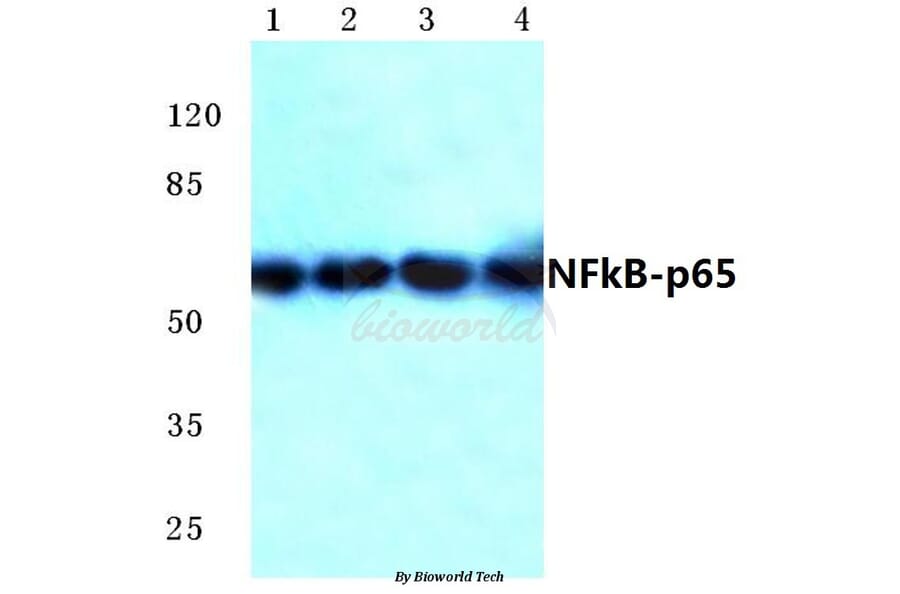
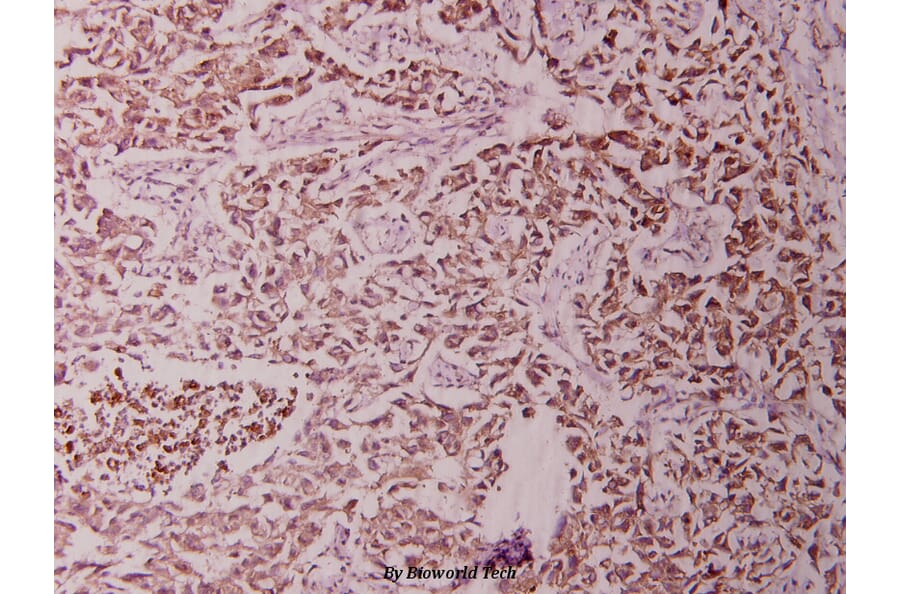
Unconjugated
Metastasis, the greatest clinical challenge associated with cancer, is closely connected to multiple biological processes, including invasion and adhesion. The hypoxic environment in tumors is an important factor that causes tumor metastasis by activating HIF-1α. Fucoidan, extracted from brown algae, is a sulfated polysaccharide and, as a novel marine biological material, has been used to treat various disorders in China, Korea, Japan and other countries. In the present study, we demonstrated that fucoidan derived from Undaria pinnatifida sporophylls significantly inhibits the hypoxia-induced expression, nuclear translocation and activity of HIF-1α, the synthesis and secretion of VEGF-C and HGF, cell invasion and lymphatic metastasis in a mouse hepatocarcinoma Hca-F cell line. Fucoidan also suppressed lymphangiogenesis in vitro and in vivo. In addition, accompanied by a reduction in the HIF-1α nuclear translocation and activity, fucoidan significantly reduced the levels of p-PI3K, p-Akt, p-mTOR, p-ERK, NF-κB, MMP-2 and MMP-9, but increased TIMP-1 levels. These results indicate strongly that the anti-metastasis and anti-lymphangiogenesis activities of fucoidan are mediated by suppressing HIF-1α/VEGF-C, which attenuates the PI3K/Akt/mTOR signaling pathways.
The study aims to investigate the effects of protocatechuic acid (PCA) separated from Chinese herbs, on acute lung injury (ALI) induced by lipopolysaccharide (LPS) in mice. The mouse model was induced by intraperitoneal injection of LPS at the dose of 5mg/kg body weight. Three doses of PCA (30, 15, 5 mg/kg) were administered to mice with intraperitoneal injection one hour prior to LPS exposure. Six hours later after LPS administration, the effect of PCA on ALI mice was assessed via histopathological examination by HE staining, inflammatory cytokine production by ELISA assay and RT-PCR, p38MAPK and NF-κB activation by Western blot analysis. We found that PCA administration significantly ameliorated lung histopathological changes and decreased protein concentration in the bronchoalveolar lavage fluid. Furthermore, the overproduction of tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) was reduced by PCA. Additionally, PCA at the dose of 30 mg/kg could block the activation of p38MAPK and NF-κB signal pathways induced by LPS. In conclusion, our findings demonstrate that PCA possesses a protective effect on LPS-induced ALI in mice via suppression of p38MAPK and NF-κB signal pathways. Therefore, PCA may be useful in the therapy of lung inflammatory diseases, especially for ALI.
CDDO-Me, initiated in a phase II clinical trial, is a potential useful therapeutic agent for cancer and inflammatory dysfunctions, whereas the therapeutic efficacy of CDDO-Me on LPS-induced acute lung injury (ALI) has not been reported as yet. The purpose of the present study was to explore the protective effect of CDDO-Me on LPS-induced ALI in mice and to investigate its possible mechanism. BalB/c mice received CDDO-Me (0.5mg/kg, 2mg/kg) or dexamethasone (5mg/kg) intraperitoneally 1h before LPS stimulation and were sacrificed 6h later. W/D ratio, lung MPO activity, number of total cells and neutrophils, pulmonary histopathology, IL-6, IL-1β, and TNF-α in the BALF were assessed. Furthermore, we estimated iNOS, IL-6, IL-1β, and TNF-α mRNA expression and NO production as well as the activation of the three main MAPKs, AkT, IκB-α and p65. Pretreatment with CDDO-Me significantly ameliorated W/D ratio, lung MPO activity, inflammatory cell infiltration, and inflammatory cytokine production in BALF from the in vivo study. Additionally, CDDO-Me had beneficial effects on the intervention for pathogenesis process at molecular, protein and transcriptional levels in vitro. These analytical results provided evidence that CDDO-Me could be a potential therapeutic candidate for treating LPS-induced ALI.
Cisplatin is a first-line chemotherapy drug against ovarian cancer. However, its strong toxic side effects and the development of cisplatin resistance in human cancer cells seriously influence the effects of chemotherapy and quality of life in patients. Noscapine (Nos), a non-toxic benzylisoquinoline alkaloid extracted from opium, has been recently reported to have anti-cancer activity, but the mechanism of that effect has not been clearly established. In the present study, we investigated cytotoxicity of Nos in combination with cisplatin (DDP) in drug-resistant human ovarian cancer cell line SKOV3/DDP in vitro and in vivo null mice xenograft model. Cell proliferation was measured by MTT assay, flow cytometry was used to analyze cell cycle and apoptosis, protein expression of several apoptotic factors was investigated by flow cytometry and immunohistochemical method, and their mRNA expression levels were determined by real-time PCR. In vitro experiments showed that Nos significantly inhibited proliferation of SKOV3/DDP cells. DDP/Nos-combined treatment notably enhanced DDP-induced inhibition of cell proliferation and increased the pro-apoptotic effect of DDP in SKOV3/DDP cells. DDP/Nos administration increased the proportion of G2/M cells, reduced both protein and mRNA expression of anti-apoptotic factors XIAP, surviving and NF-kB, and augmented protein and mRNA levels of pro-apoptotic caspase-3. In vivo experiments revealed that Nos/DDP treatment increased the apoptotic rate of xenograft tumors in null mice. Tumor volume decreased from 1.733 ± 0.155 g in mice treated with DDP alone to 1.191 ± 0.106 g in animals treated with Nos/DDP. These observations suggest that Nos increases the anti-cancer activity of DDP against the drug-resistant ovarian cancer cell line SKOV3/DDP by modulating the cell cycle and activating apoptotic pathways. The study provides a new chemotherapy strategy for the treatment of DDP-resistant human ovarian cancer.
Tumor-stroma interactions are referred to as essential events in tumor progression. There has been growing attention that bone marrow-derived mesenchymal stem cells (BMSCs) can travel to tumor stroma, where they differentiate into tumor-associated fibroblast (TAF)-like cells, a predominant tumor-promoting stromal cell. However, little is definitively known about the contributors for this transition. Here, using an in vitro direct co-culture model of colon cancer cells and BMSCs, we identify that colon cancer cells can induce adjoining BMSCs to exhibit the typical characteristic of TAFs, with increased expression of α-smooth muscle actin (α-SMA). Importantly, the present data also reveals that activated Notch signaling mediates transformation of BMSCs to TAFs through the downstream TGF-β/Smad signaling pathway.
Mitofusin 2 (Mfn2) is a dynamin-like protein anchored in the outer mitochondrial membrane that plays a crucial role in ensuring optimal mitochondrial morphological homeostasis. It has been shown that reduced expression of Mfn2 is associated with insulin resistance, but the mechanism is still unclear. We investigated whether Mfn2 deficiency leads to impaired insulin sensitivity via elevated oxidative stress. L6 skeletal muscle cells were treated with palmitate and Mfn2 expression was repressed by transfection with antisense Mfn2. Levels of antioxidant enzymes, reactive oxygen species (ROS), the phosphorylation of c-Jun N-terminal Kinase (JNK) and nuclear factor-κB (NF-κB) and the mitochondrial membrane potential (Δψm) were measured. The results showed palmitate-induced insulin resistance of skeletal muscle cells was accompanied by Mfn2 repression. Meanwhile, the cells had decreased Δψm and activity of antioxidant enzymes which could increase production of ROS, phosphorylation of JNK and NF-κB. When Mfn2 was up-regulated in palmitate-treated cells, oxidative stress and insulin resistance was alleviated. Furthermore, knock-down of Mfn2 in control cells enhanced oxidative stress. Mfn2 deficiency led to increased superoxide concentration and activation of JNK as well as NF-κB associated with insulin signaling. In conclusion, Mfn2 is a potent repressor for oxidative stress and regulation of Mfn2 expression may prove to be a potential method to circumvent insulin resistance.
This study aimed to assess BA impact on inflammation markers and repair of intestinal mucosa. Forty-eight rats were randomly divided into stress (n = 24) and BA (n = 24) groups. Stress was induced by fettering in all animals, fed enterally with 125.4 kJ/kg/d and 0.2 g/kg/d nitrogen. Then, rats were treated for 8 days with 5 mg/kg/d BA (BA group) or 5 mg/kg/d saline (Stress group). Levels of NF-κB, IL-10, TNF-α, and IFN-γ were measured at different time points, in plasma and intestinal mucosa samples. Changes in intestinal mucosa morphology were observed by electron microscopy. Plasma and/or mucosal levels of NF-κB, TNF-α, and IFN-γ were significantly higher in both groups after stress induction (P < 0.05). These high levels persisted in control animals throughout the experiment, and were significantly reduced in the BA group, 3 and 8 days after stress induction (P < 0.05). Interestingly, IL-10 levels were increased after BA treatment (P < 0.05). At day 8, ileal mucosal villi and crypt structure were significantly restored in the BA group. Bifidobacterial adhesin plays a role in repairing intestinal mucosa injury after stress by regulating the release of inflammatory mediators in the intestinal mucosa.
Cichoric acid extract (CAE) from Echinacea purpurea L. was used to investigate the anti-arthritic effect by using collagen-induced arthritis (CIA) rat model. The hind paw swelling volume and the body weight were measured and recorded. All the drug solutions were administered orally to rats for a total of 28 days. On day 28, the rats were anaesthetized and decapitated. The thymus and spleen were weighed for the determination of the organ index. The concentration of tumor necrosis factor alpha (TNFα), interleukin-1 beta (IL-1β) and prostaglandin E2 (PGE-2) in the serum was measured using commercially available ELISA kits. Total and phosphor-NF-κB and Cox-2 protein expression in synovial tissues were determined by histological slides quantification and western blot analysis. Our data showed that administration of all doses of CAE (8, 16, and 32 mg/kg) significantly decreased the paw swelling, restored body weight gain and decreased the organ index of the thymus and spleen compared with that of the CIA group. CAE (8, 16, and 32 mg/kg) treatment significantly reduced the levels of TNFα, IL-1β and PGE-2 in serum compared with the CIA group. Histopathological analysis demonstrated that CAE has obvious anti-arthritic activity. In addition, CAE (32 mg/kg) significantly decreased the levels of nuclear factor-κB (NF-κB), TNFα and cyclooxygenase 2 (Cox-2) in synovium tissues of the ankle joint compared with the CIA group. Furthermore, CAE administration significantly decreased the protein expression of phosphor-NF-κB and Cox-2 in synovium tissues of the knee joint compared with the CIA group. The results suggest that the anti-inflammatory activity of CAE may account for its anti-arthritic effect, and CAE could be a potential therapeutic drug for the treatment of rheumatoid arthritis (RA).
Angiogenin (ANG) is reportedly multifunctional, with roles in angiogenesis and autoimmune diseases. This protein is involved in the innate immune system and has been implicated in several inflammatory diseases. Although ANG may be involved in the anti-inflammatory response, there is no evidence that it has direct anti-inflammatory effects. In this study we sought to determine whether ANG has an anti-inflammatory effect in human corneal fibroblasts (HCFs) exposed to media containing tumor necrosis factor-alpha (TNF-α). We found that ANG reduced the mRNA expression of interleukin-1 beta (IL-1β), -6, -8 and TNF-α receptors (TNFR) 1 and 2. In contrast, ANG increased the mRNA expression of IL-4 and -10. Protein levels of TANK-binding kinase 1 (TBK1) were reduced by ANG in HCFs treated with TNF-α. Moreover, ANG diminished the expression of IL-6 and -8 and monocyte chemotactic protein- (MCP-) 1. The protein expression of nuclear factor-κB (NF-κB) was downregulated by ANG treatment. These findings suggest that ANG suppressed the TNF-α-induced inflammatory response in HCFs through inhibition of TBK1-mediated NF-κB nuclear translocation. These novel results are likely to play a significant role in the selection of immune-mediated inflammatory therapeutic targets and may shed light on the pathogenesis of immune-mediated inflammatory diseases.
Accumulating evidence indicate that macrophages activate mesenchymal stem cells (MSCs) to acquire pro-inflammatory phenotype. However, the role of MSCs activated by macrophages in gastric cancer remains largely unknown. In this study, we found that MSCs were activated by macrophages to produce increased levels of inflammatory cytokines. Cell colony formation and transwell migration assays revealed that supernatants from the activated MSCs could promote both gastric epithelial cell and gastric cancer cell proliferation and migration. In addition, the expression of epithelial-mesenchymal transition (EMT), angiogenesis, and stemness-related genes was increased in activated MSCs. The phosphorylated forms of NF-κB, ERK and STAT3 in gastric cells were increased by active MSCs. Inhibition of NF-κB activation by PDTC blocked the effect of activated MSCs on gastric cancer cells. Co-injection of activated MSCs with gastric cancer cells could accelerate gastric cancer growth. Moreover, human peripheral blood monocytes derived macrophages also activated MSCs to prompt gastric cancer cell proliferation and migration. Taken together, our findings suggest that MSCs activated by macrophage acquire pro-inflammatory phenotype and prompt gastric cancer growth in an NF-κB-dependent manner, which provides new evidence for the modulation of MSCs by tumor microenvironment and further insight to the role of stromal cells in gastric carcinogenesis and cancer progression.
Blood-brain barrier (BBB) disruption and brain edema formation play important roles in the secondary neuronal death and neurological dysfunction induced by intracerebral hemorrhage (ICH). Poloxamer 188 (P188), a multiblock copolymer surfactant, has been shown to be capable of sealing damaged cell membranes and decrease neuronal cell death. In this study, we explored whether P188 had a protective effect against ICH and its underlying mechanisms. Male ICR mice were subjected to infusion of type IV collagenase (to induce ICH) of saline (for shams) into the left striatum. The results showed that P188-12 mg post-treatment by tail intravenous injection significantly ameliorated the neurological symptoms and brain edema, attenuated BBB permeability, and decreased cell insults and injury volume at 24 and 72 h after ICH. Furthermore, P188 maintained the protein levels of tight junction (TJ) proteins including claudin-5, occludin, and zonula occludens-1, and reversed the increases of nuclear factor-kappaB (NF-κB), matrix metalloproteinase (MMP)-2, and MMP-9 protein expression at 72 h post ICH. Immunofluorescence showed P188 treatment rearranged the structure of TJ proteins in a continuous and linear pattern. Therefore, the present study concludes that P188 can protect against ICH, and the protective effect was associated with preventing BBB disruption through NF-κB-MMPs-mediated TJ proteins degradation.
D-galactosamine (GalN)/lipopolysaccharide (LPS)-induced lethality and acute liver failure is dependent on endogenously produced inflammatory cytokines. Adenosine has been proven to be a central role in the regulation of inflammatory response. It is not entirely clear that which adenosine action is actually crucial to limiting inflammatory tissue destruction. Here we showed that GalN/LPS challenge elevated hepatic adenosine and induced lethality in adenosine receptor-deficient mice with equal efficiency as wild-type mice. In GalN/LPS-treated mice, pretreatment with adenosine 5'-monophosphate (5'-AMP) significantly elevated hepatic adenosine level and reduced mortality through decreasing cytokine and chemokine production. In RAW264.7 cells, 5'-AMP treatment inhibited the production of inflammatory cytokines, which is not mediated through adenosine receptors. 5'-AMP failed to attenuate LPS-induced nuclear factor-κB (NF-κB) p65 nuclear translocation, but reduced LPS-induced recruitment of NF-κB p65 to inflammatory gene promoters and decreased LPS-induced enrichment of H3K4 dimethylation at the tumor necrosis factor-α (TNF-α) promoter, which was involved in 5'-AMP-induced elevation of cellular adenosine and a decline of methylation potential. In vitro biochemical analysis revealed that adenosine directly attenuated recruitment of NF-κB to the TNF-α and interleukin-6 promoters. Our findings demonstrate that 5'-AMP-inhibiting inflammatory response is not mediated by adenosine receptors and it may represent a potential protective agent for amelioration of LPS-induced liver injury.
Aldose reductase (AR) has a key role in several inflammatory diseases: diabetes, cancer and cardiovascular diseases. Therefore, AR inhibition seems to be a useful strategy for anti-inflammation therapy. In the central nervous system (CNS), microglial over-activation is considered to be a central event in neuroinflammation. However, the effects of AR inhibition in CNS inflammation and its underlying mechanism of action remain unknown. In the present study, we found that FMHM (a naturally derived AR inhibitor from the roots of Polygala tricornis Gagnep.) showed potent anti-neuroinflammatory effects in vivo and in vitro by inhibiting microglial activation and expression of inflammatory mediators. Mechanistic studies showed that FMHM suppressed the activity of AR-dependent phospholipase C/protein kinase C signaling, which further resulted in downstream inactivation of the IκB kinase/IκB/nuclear factor-kappa B (NF-κB) inflammatory pathway. Therefore, AR inhibition-dependent NF-κB inactivation negatively regulated the transcription and expression of various inflammatory genes. AR inhibition by FMHM exerted neuroprotective effects in lipopolysaccharide-induced neuron-microglia co-cultures. These findings suggested that AR is a potential target for neuroinflammation inhibition and that FMHM could be an effective agent for treating or preventing neuroinflammatory diseases.
Lipoxins (LXs) and their analogues are known to display potent anti-inflammatory actions. Previously, we reported that lipoxin A4 (LXA4) possessed powerful anti-inflammatory properties in acute pancreatitis in rats and that it may ameliorate the concomitant acute lung injury by reducing cytokine generation and inhibiting neutrophil activation. Considering that the vascular endothelium plays an important role during adherence, migration and activation of leukocytes, the present study was designed to investigate the effects of LXA4 on the inflammatory response induced by tumor necrosis factor α (TNF-α) in human pulmonary microvascular endothelial cells (HPMECs) and explore the potential mechanisms involved in these processes. We found that LXA4 markedly down-regulated the expression of monocyte chemotactic protein-1 (MCP-1), E-selectin, and interleukin-6 (IL-6) mRNA, as well as intercellular adhesion molecule-1 (ICAM-1) in TNF-α-exposed HPMECs. Moreover, LXA4 inhibited the phosphorylation and nuclear translocation of nuclear factor-κB/p65 (NF-κB/p65) and phosphorylation of p38 mitogen-activated protein kinase (p38 MAPK) in HPMECs following TNF-α stimulation. Heme oxygenase-1 (HO-1), a cytoprotective enzyme, was up-regulated by LXA4 in both non- and TNF-α-stimulated HPMECs. In conclusion, the protective effects of LXA4 to ALI may be executed through inhibition inflammation pathways of NF-κB and p38 MAPK and up-regulation of cytoprotective HO-1.
Artemisinin and its derivatives have been reported to have immunosuppressive activity in some laboratory studies. However, the detail of mechanism remains to be demonstrated. The objective of this study is to clarify the immunosuppressive activity of artesunate (AST), one kind of artemisinin derivatives, and to find its unexplored mode of action. In vitro, the proliferation of T lymphocytes and its cytotoxicity were measured by WST-1 and MTT assay. In vivo, the immunomodulatory effect of AST was evaluated in a mouse model of delayed type hypersensitivity reaction (DTH), which was based on a T cell-mediated immune response. The data displayed that AST had a relatively high immunosuppressive activity with low toxicity, and could inhibit T lymphocyte proliferation induced by mitogen and alloantigen. Meanwhile, topical administration of AST could suppress DTH response significantly. Moreover, AST could also increase the secretion of TFG-β, coupling with the striking enhance of NF-κB/p65 and Smad2/3 signaling. The promotion of CD4(+)CD25(+) regulatory T cells (Tregs) was shown to be a possible mechanism involved in AST-mediated regulation. Taken together, these observations exhibit the potential of developing AST as a novel safe remedy for the treatment of T cell-mediated immune disorders.
Controversial roles of FOXP3 in different cancers have been reported previously, while its role in gastric cancer is largely unknown. Here we found that FOXP3 is unexpectedly upregulated in some gastric cancer cells. To test whether increased FOXP3 remains the tumor suppressor role in gastric cancer as seen in other cancers, we test its function in cell proliferation both at basal and TNFα mimicked inflammatory condition. Compared with the proliferation inhibitory role observed in basal condition, FOXP3 is insufficient to inhibit the cell proliferation under TNFα treatment. Molecularly, we found that TNFα induced an interaction between FOXP3 and p65, which in turn drive the FOXP3 away from the promoter of the well known target p21. Our data here suggest that although FOXP3 is upregulated in gastric cancer, its tumor suppressor role has been dampened due to the inflammation environment.
Receptor for advanced glycation end products (RAGE) is associated with inflammation and the progression of cardiovascular diseases. The current study tested the hypothesis that RAGE is involved in the pathogenesis of aortic valve (AV) calcification. Pioglitazone attenuated AV calcification in experimental hypercholesterolemic rabbits via down-regulation of RAGE. Male New Zealand rabbits weighing 2.5-3.0 kg were randomly divided into three groups: control group, high cholesterol + vitamin D(2) (HC + vitD(2)) group and HC + vitD(2) supplemented with pioglitazone group. Compared with HC + vitD(2) group, pioglitazone significantly inhibited the progression of AV calcification assessed by echocardiography. HC + vitD(2) diet markedly increased RAGE expression, oxidative stress, inflammatory cells infiltration and osteopontin expression. These changes were also significantly attenuated by administration of pioglitazone. Cultured porcine aortic valve interstitial cells (VICs) were used as in vitro model. We found that advanced glycation end products of bovine serum albumin markedly increased the expression of RAGE, induced high levels of production of pro-inflammatory cytokines and promoted osteoblastic differentiation of VICs. However, these effects were found to be remarkably suppressed by siRNA silencing of RAGE and pioglitazone as well. Our data provide evidence that RAGE activation-induced inflammation promotes AV calcification in hypercholesterolemic rabbits, which can be attenuated by pioglitazone treatment. This beneficial effect is associated with remarkable down-regulation of RAGE expression.
Neonates with intrauterine growth retardation (IUGR) often suffer from impaired cellular immunity, and weaning may further aggravate adverse effects of IUGR on development and function of the immune system. In this study, we investigated effects of glutamine supplementation on immune status in the intestines of weaning pigs with IUGR, focusing on molecular mechanisms underlying altered immune response. Piglets with IUGR were weaned at 21 days of age and received orally 1.22 g alanine or 1 g glutamine per kg body weight every 12 h. Weight gain and intestinal weight of weaning piglets were increased by glutamine supplementation. Levels of serum IgG in piglets supplemented with glutamine were increased compared with Control piglets. The production of IL-1 and IL-8 in the serum and jejunum was decreased by glutamine supplementation, whereas the levels of IL-4 in the serum and the concentrations of IL-4 and IL-10 in the jejunum were increased. The expression of heat shock protein 70 (Hsp70) in the jejunum was increased by glutamine supplementation, but the degradation of inhibitor κB and the activity of nuclear factor-κB (NF-κB) were decreased. In conclusion, glutamine supplementation enhanced immune response in weaning piglets with IUGR. The effects of glutamine in IUGR are associated with increased Hsp70 expression and suppression of NF-κB activation.
Lung carcinogenesis is a complex process in an unregulated inflammatory environment. Curcumin has been extensively investigated as a multi-target anti-tumor and anti-inflammation compound. In this paper, we demonstrate a novel inflammation-related mechanism for curcumin-induced inhibition of lung tumor growth. We found that neutrophil elastase, an important regulator of inflammatory processes, directly triggered tumor cell proliferation in human lung adenocarcinoma A549 cells, and curcumin could completely suppress the excess tumor proliferation induced by neutrophil elastase. α1-antitrypsin is synthesized by tumor cells and is the natural inhibitor of neutrophil elastase. We found that curcumin counteracted the decrease of α1-antitrypsin induced by neutrophil elastase by inducing the promoter activity of α1-antitrypsin and promoting its expression in A549 cells. The inhibition of neutrophil elastase-induced proliferation by curcumin was dependent on the PI3K/Akt pathway. Knockdown of α1-antitrypsin by siRNA further enhanced the tumor cell proliferation induced by neutrophil elastase and significantly blocked the anti-proliferation effect of curcumin against neutrophil elastase. Curcumin remarkably inhibited the primary tumor growth of Lewis lung carcinoma (LLC) in C57BL/6 mice. We further showed that curcumin upregulated the level of α1-antitrypsin in primary tumor tissue by promoting its local expression, and the protein level of neutrophil elastase in tumor tissue was obviously decreased in mice treated with curcumin. Overall, our results suggest that neutrophil elastase and α1-antitrypsin play important roles in modulating lung tumor proliferation in inflammatory microenvironment and curcumin inhibits neutrophil elastase-induced tumor proliferation via upregulating α1-antitrypsin expression in vitro and in vivo.
Hydrogen sulfide (H(2)S) has been shown to protect against oxidative stress injury and inflammation in various hypoxia-induced insult models. However, it remains unknown whether H(2)S protects human skin keratinocytes (HaCaT cells) against chemical hypoxia-induced damage. In the current study, HaCaT cells were treated with cobalt chloride (CoCl(2)), a well known hypoxia mimetic agent, to establish a chemical hypoxia-induced cell injury model. Our findings showed that pretreatment of HaCaT cells with NaHS (a donor of H(2)S) for 30 min before exposure to CoCl(2) for 24 h significantly attenuated CoCl(2)-induced injuries and inflammatory responses, evidenced by increases in cell viability and GSH level and decreases in ROS generation and secretions of IL-1β, IL-6 and IL-8. In addition, pretreatment with NaHS markedly reduced CoCl(2)-induced COX-2 overexpression and PGE(2) secretion as well as intranuclear NF-κB p65 subunit accumulation (the central step of NF-κB activation). Similar to the protective effect of H(2)S, both NS-398 (a selective COX-2 inhibitor) and PDTC (a selective NF-κB inhibitor) depressed not only CoCl(2)-induced cytotoxicity, but also the secretions of IL-1β, IL-6 and IL-8. Importantly, PDTC obviously attenuated overexpression of COX-2 induced by CoCl(2). Notably, NAC, a ROS scavenger, conferred a similar protective effect of H(2)S against CoCl(2)-induced insults and inflammatory responses. Taken together, the findings of the present study have demonstrated for the first time that H(2)S protects HaCaT cells against CoCl(2)-induced injuries and inflammatory responses through inhibition of ROS-activated NF-κB/COX-2 pathway.
Oxidative stress is a major cause in neurodegenerative diseases including Alzheimer's disease (AD), Parkinson's disease (PD), and cerebral ischemia. Ginsenoside Rg1, a natural product extracted from Panax ginseng C.A. Meyer, has been reported to exert notable neuroprotective activities, which partly ascribed to its antioxidative activity. However, its molecular mechanism against oxidative stress induced by exogenous hydrogen peroxide (H(2)O(2)) remained unclear. In this study, we investigated its effect on H(2)O(2)-induced cell death and explored possible signaling pathway in PC12 cells. We proved that pretreatment with Rg1 at concentrations of 0.1-10 μM remarkably reduced the cytotoxicity induced by 400 μM of H(2)O(2) in PC12 cells by MTT and Hoechst and PI double staining assay. Of note, we demonstrated the activation of NF-κB signaling pathway induced by H(2)O(2) thoroughly in PC12 cells, and Rg1 suppressed phosphorylation and nuclear translocation of NF-κB/p65, phosphorylation and degradation of inhibitor protein of κB (IκB) as well as the phosphorylation of IκB-kinase complex (IKK) by western blotting or indirect immunofluorescence assay. Besides, Rg1 also inhibited the activation of Akt and the extracellular signal-regulated kinase 1/2 (ERK1/2). Furthermore, the protection of Rg1 on H(2)O(2)-injured PC12 cells was attenuated by pretreatment with two NF-κB pathway inhibitors (JSH-23 or BOT-64). In conclusion, our results suggest that Rg1 could rescue the cell injury by H(2)O(2) via down-regulation NF-κB signaling pathway as well as Akt and ERK1/2 activation, which put new evidence on the neuroprotective mechanism of Rg1 against the oxidative stress and the regulatory role of H(2)O(2) in NF-κB pathway in PC12 cells.
The acid polysaccharide fraction (APSF) extracted from the mycelia of cultivated Cordyceps sinensis is water-soluble polysaccharide. In this study we evaluated the modulating effects of APSF on murine macrophage cell line RAW264.7. Phagocytotic assay by neutral red and FITC-dextran internalization showed that APSF stimulated the phagocytosis of macrophages. The nitrite levels in the culture supernatant determined using Griess reagent revealed the elevation of NO production after treatment with APSF. RT-PCR and immunocytochemistry assay indicated that APSF promoted both the mRNA and protein expressions of inducible nitric oxide synthase (iNOS). Furthermore, Western blotting demonstrated that NF-kappaB levels in nucleuses increased after APSF treatment, suggesting that APSF probably stimulated macrophage activities by activating the IkappaB-NF-kappaB pathway.
BACKGROUND/AIMS:
Age-associated and stress-induced involution of the thymus is accompanied by reduced numbers of thymic epithelial cells (TECs) and severe reduction in peripheral T cell repertoire specificities. These events seriously affect immune function, but the mechanisms involved are unclear. Our preliminary findings showed that doxycycline (Dox) could drive the proliferation of a TEC line (MTEC1 cells) partially via the MAPK signaling pathway. Dox can also up-regulate IL-6 and GM-CSF expression via the NF-κB and MAPK/ERK pathways. Herein, we investigate the effects and mechanisms used by Dox that protect against mitomycin C (MMC)-induced MTEC1 cell apoptosis.
METHODS:
MTEC1 cells were treated with Dox, MMC, and Dox plus MMC for different amounts of time. The expression of Trx2, NF-κB, Bcl-2, and Bax proteins were then detected by western blotting.
RESULTS:
Our findings show that Dox protects MTEC1 cells from MMC-induced apoptosis. Dox up-regulated the expression of Trx2 and promoted NF-κB phosphorylation. Meanwhile, Dox also increased the expression of Bcl-2, partially reduced the expression of Bax, and normalized the ratio of Bcl-2 to Bax.
CONCLUSION:
Dox exerts an anti-apoptosis function via the NF-κB-Bcl-2/Bax and Trx2-ASK1/JNK pathways in vitro. Therefore, Dox may represent a drug that could be used to attenuate thymic senescence, rescue thymic function, and promote T cell reconstitution.
© 2016 The Author(s) Published by S. Karger AG, Basel.
BACKGROUND:
Resveratrol is a phytoalexin with beneficial effects on human health. The aim of the present study was to investigate the effects of resveratrol on endothelial dysfunction involved in insulin signaling and inflammation.
METHODS:
Endothelial cells were stimulated with palmitate (PA) to induce insulin resistance characterized by a loss of insulin-mediated nitric oxide (NO) production. Diabetes was induced in rats by fructose feeding. The effects of resveratrol and the mechanisms involved were investigated using an aortic relaxation assay and Western blot analysis.
RESULTS:
In endothelial cells, 0.1-10 μmol/L resveratrol suppressed IκB kinase β (IKKβ)/nuclear factor-κB phosphorylation, as well as tumor necrosis factor-α and interleukin-6 production, and restored the insulin receptor substrate-1 (Irs-1)/Akt/endothelial NO synthase signaling pathway. Furthermore, resveratrol effectively inhibited the mitogenic actions of insulin by decreasing the secretion of endothelin-1 and plasminogen activator inhibitor-1. It also positively regulated AMP-activated kinase (AMPK) and sirtuin 1 (SIRT1) activation, which contributed to the inhibition of inflammation implicated in endothelial insulin resistance. Stimulation with PA and long term-fructose feeding impaired insulin-mediated vessel dilation in rat aorta, whereas pretreatment of aortic rings with resveratrol (0.1-10 μmol/L) or treatment of rats with 5 or 20 mg/kg resveratrol counteracted these changes.
CONCLUSION:
The results indicate that resveratrol inhibits inflammation and facilitates insulin phosphatidylinositol 3-kinase signaling by beneficial modulation of IRS-1 function partly via regulation of AMPK and SIRT1 activity in the endothelium.
© 2015 Ruijin Hospital, Shanghai Jiaotong University School of Medicine and John Wiley Sons & Australia, Ltd.
AIMS:
Altered drug disposition has been associated with inflammation and diabetes, leading to the alteration of drug efficacy and toxicity. Carboxylesterases are major hydrolytic enzymes in the liver, catalyzing the hydrolytic biotransformation of numerous therapeutic agents. Therefore, how glucose affects the regulation of carboxylesterases by interleukin-6 (IL-6) and lipopolysaccharide (LPS) were investigated.
MAIN METHODS:
Primary mouse hepatocytes were cultured. Protein levels were measured by Western blot or enzyme linked immunosorbent assay (ELISA), while confocal laser scanning microscope and flow cytometry were used to confirm the activation of pregnane X receptor (PXR). Carboxylesterase activity was evaluated by enzymatic and toxicological assays.
KEY FINDINGS:
Elevated glucose (11 or 25 mM) significantly increased carboxylesterase expression compared to 5.6 mM glucose. Carboxylesterase expression and activity were inhibited by LPS or IL-6 in 25 mM glucose, but stimulated in 5.6 mM glucose. The altered expression of carboxylesterases was not consistent with the activation of nuclear factor kappa B (NFκB) but repeatedly with the expression and activation of pregnane X receptor (PXR). The altered activation of PXR was further evidenced by the differential subcellular translocation and the expression of its target gene multidrug resistance 1 (MDR1). It implies that PXR, instead of inflammatory signaling, mediates the regulation of carboxylesterases by inflammatory mediators in different glucose concentrations.
SIGNIFICANCE:
The findings contribute to clarify the regulation of carboxylesterases by inflammatory mediators, and indicate that carboxylesterase-involved drug metabolism and drug-drug interactions in diabetes should be reevaluated according to the intensity of inflammatory reactions and hyperglycemia.
Copyright © 2014 Elsevier Inc. All rights reserved.
BACKGROUND/AIMS:
Colorectal carcinoma is one of the most common cancers world-wide, with high morbidity and mortality rates. Arginine ADP-ribosyltransferase 1(ART1) is an important ecto-ADP-ribose transferase and has been proven to be intimately involved in a number of biological processes. However, the influence of ART1 on survival and apoptosis of colorectal carcinoma cells and the potential mechanism of action of ART1 remain uncharacterized.
METHODS:
ART1 was silenced via lentiviral vector-mediated short hairpin RNA (shRNA) in CT26 colon carcinoma cells, and cisplatin (CDDP) was applied to induce apoptosis. Survival and apoptosis rate of CT26 cells was assessed by CCK8 assay, flow cytometry and Hoechst 33342 staining. Expression and activity of signaling proteins were detected by Western blot.
RESULTS:
ART1 knockdown enhanced the inhibition of cell survival and increased the apoptosis induced by CDDP. Furthermore, the reduced survival rate correlated with reduced levels of phos-Akt(Thr308) and phos-IκBα and reduced NF-κB p65 nuclear translocation. A decline in Bcl-2 and Bcl-xl expression and an increase in Bax expression may explain the enhanced apoptosis.
CONCLUSION:
This study provides a molecular mechanism for the function of ART1 in colorectal carcinoma and defines a potential therapeutic target for the enhanced treatment of this prominent world-wide disease.
© 2013 S. Karger AG, Basel.
AIMS/HYPOTHESIS:
In type 2 diabetes mellitus (T2DM), the abnormal protein and lipid composition of diabetic high-density lipoprotein (HDL) could impair its anti-inflammatory functions. Whether nonenzymatic glycation directly impaired the anti-inflammatory effects of HDL in innate immunity remained unclear.
METHODS:
Human acute monocytic leukemia cell line (THP-1) cells, mouse RAW 264.7 macrophages and primary human monocytes derived macrophages were pre-incubated with native HDL, diabetic HDL isolated from T2DM patients or HDL glycated with different doses of d-glucose in vitro and then challenged with lipopolysaccharide (LPS). The release of tumor necrosis factor (TNF)-α and IL-1β was assayed by enzyme-linked immunosorbent assay (ELISA). Phosphorylation of Iκ-Bα in cytoplasm and nuclear translocation of NF-κB were detected by western blot. Glycation levels of native HDL, glycated HDL and diabetic HDL were determined using LC-MS/MS.
RESULTS:
The potency of diabetic HDL to inhibit the release of TNF-α (p < 0.05) and IL-1β (p < 0.001) was dramatically attenuated compared with that of native HDL. Similarly, glycation of HDL in vitro impaired its ability to inhibit TNF-α and IL-1β release in a glucose dose-dependent manner. Moreover, apoHDL still effectively inhibited the release of TNF-α and IL-1β induced by LPS, but glycated apoHDL partly lost such abilities. Nonenzymatic glycation levels of glycated HDL and diabetic HDL increased 28 fold (p < 0.001) and 4 fold (p < 0.001), respectively compared with that of native HDL.
CONCLUSIONS:
In this study, we observed that diabetic HDL and HDL glycated in vitro both partly lose their protective effects to inhibit cytokines release induced by LPS in macrophages, and nonenzymatic glycation of the protein components of HDL plays key roles in these impairments.
Copyright © 2011 John Wiley & Sons, Ltd.
BACKGROUND:
Most patients after liver transplantation (LT) suffer from intestinal barrier dysfunction. Glycyl-glutamine (Gly-Gln) by parenteral supplementation is hydrolyzed to release glutamine, which improves intestinal barrier function in intestinal injury. This study aimed to investigate the effect of Gly-Gln by enteral supplementation on intestinal barrier function in rats after allogenetic LT under immunosuppressive therapy.
METHODS:
Twelve inbred Lewis rats were selected randomly as donors, and 24 inbred Brown Norway (BN) rats as recipients of allogenetic LT. The recipients were divided into a control group (Ala, n=12) and an experimental group (Gly-Gln, n=12). In each group, 6 normal BN rats were sampled for normal parameters on preoperative day 3. The 6 recipients in the control group received alanine (Ala) daily by gastric perfusion for 3 preoperative days and 7 postoperative days, and the 6 recipients in the experimental group were given Gly-Gln in the same manner. The 12 BN recipients underwent orthotopic LT under sterile conditions after a 3-day fast and were given immunosuppressive therapy for 7 days. They were harvested for sampling on postoperative day 8. The following parameters were assessed: intestinal mucosal protein content, mucosal ultrastructure, ileocecal sIgA content, portal plasma levels of endotoxin and TNF-alpha, and bacterial translocation.
RESULTS:
All recipients were alive after LT. On preoperative day 3, all parameters were similar in the two groups. On postoperative day 8, all parameters in the two groups were remarkably changed from those on preoperative day 3. However, compared to the Ala group, supplementation with Gly-Gln increased the levels of intestinal mucosal protein and ileocecal sIgA, improved mucosal microvilli, and decreased portal plasma levels of endotoxin and TNF-alpha as well as bacterial translocation.
CONCLUSION:
Enteral supplementation with Gly-Gln improved intestinal barrier function after allogenetic LT in rats.