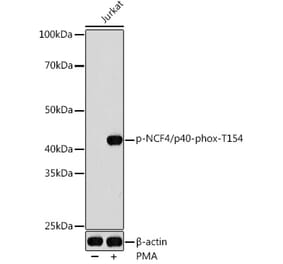
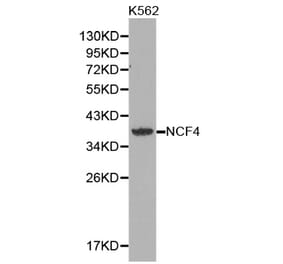
Unconjugated
Transforming growth factor-β1 (TGF-β1) induces expression of the proinflammatory and profibrotic cytokine monocyte chemoattractant protein-1 (MCP-1) in tubular epithelial cells (TECs) and thereby contributes to the tubular epithelial-mesenchymal transition (EMT), which in turn leads to the progression of tubulointerstitial inflammation into tubulointerstitial fibrosis. Exactly how TGF-β1 causes MCP-1 overexpression and subsequent EMT is not well understood. Using human tubular epithelial cultures, we found that TGF-β1 upregulated the expression of reduced nicotinamide adenine dinucleotide phosphate oxidases 2 and 4 and their regulatory subunits, inducing the production of reactive oxygen species. These reactive species activated a signaling pathway mediated by extracellular signal-regulated kinase (ERK1/2) and nuclear factor-κB (NF-κB), which upregulated expression of MCP-1. Incubating cultures with TGF-β1 was sufficient to induce hallmarks of EMT, such as downregulation of epithelial marker proteins (E-cadherin and zonula occludens-1), induction of mesenchymal marker proteins (α-smooth muscle actin, fibronectin, and vimentin), and elevated cell migration and invasion in an EMT-like manner. Overexpressing MCP-1 in cells exposed to TGF-β1 exacerbated these EMT-like changes. Pretreating cells with the antioxidant and anti-inflammatory compound arctigenin (ATG) protected them against these TGF-β1-induced EMT-like changes; the compound worked by inhibiting the ROS/ERK1/2/NF-κB pathway to decrease MCP-1 upregulation. These findings suggest ATG as a new therapeutic candidate to inhibit or even reverse tubular EMT-like changes during progression to tubulointerstitial fibrosis, and they provide the first clues to how ATG may work.
A long-term "memory" of hyperglycemic stress, even when glycemia is normalized, has been previously reported in diabetes. In this report we propose a similar hypothesis that exposure to continuous high angiotensin II (Ang II) results in a cellular "memory" in isolated cardiomyocytes and in the heart tissues, and we investigate the role of NADPH oxidases in this phenomenon. Continuous high Ang II for 3 days markedly increased cardiomyocyte size, TUNEL-positive apoptotic cardiomyocytes, expression of inflammatory cytokines, and oxidative stress. These deleterious effects were also observed in the memory condition (high Ang II for 2 days followed by normal medium for 1 day). Furthermore, in a mouse model, Ang II infusion for 3 weeks significantly increased cardiac hypertrophy, apoptosis, inflammation, and ROS generation but decreased cardiac function compared with control mice, and similar effects were also observed in mice in the memory condition. Importantly, blockade of NADPH oxidase using apocynin diminished the induction of high Ang II stress markers in isolated cardiomyocytes and in the mouse heart. These effects were associated with inhibition of NADPH oxidase-mediated AKT/mTOR/S6K and ERK signaling pathways. The present results demonstrate the hypothesis that exposure to continuous high Ang II results in a hypertensive cellular memory that remains, even when cells or mice are switched back to normal Ang II. This phenomenon was associated with NADPH oxidase-mediated oxidative stress.




![Western Blot - Anti-p40-phox Antibody [ARC2553] (A306699) - Antibodies.com](https://cdn.antibodies.com/image/catalog/306/A306699_1.jpg?profile=product_alternative)