

Unconjugated
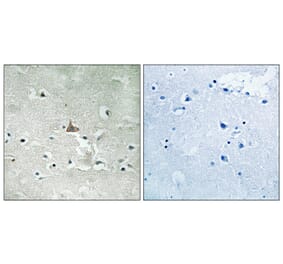
Loss of neurotrophic support in the striatum caused by reduced brain-derived neurotrophic factor (BDNF) levels plays a critical role in Huntington's disease (HD) pathogenesis. BDNF acts via TrkB and p75 neurotrophin receptors (NTR), and restoring its signaling is a prime target for HD therapeutics. Here we sought to determine whether a small molecule ligand, LM22A-4, specific for TrkB and without effects on p75(NTR), could alleviate HD-related pathology in R6/2 and BACHD mouse models of HD. LM22A-4 was administered to R6/2 mice once daily (5-6 d/week) from 4 to 11 weeks of age via intraperitoneal and intranasal routes simultaneously to maximize brain levels. The ligand reached levels in the R6/2 forebrain greater than the maximal neuroprotective dose in vitro and corrected deficits in activation of striatal TrkB and its key signaling intermediates AKT, PLCγ, and CREB. Ligand-induced TrkB activation was associated with a reduction in HD pathologies in the striatum including decreased DARPP-32 levels, neurite degeneration of parvalbumin-containing interneurons, inflammation, and intranuclear huntingtin aggregates. Aggregates were also reduced in the cortex. Notably, LM22A-4 prevented deficits in dendritic spine density of medium spiny neurons. Moreover, R6/2 mice given LM22A-4 demonstrated improved downward climbing and grip strength compared with those given vehicle, though these groups had comparable rotarod performances and survival times. In BACHD mice, long-term LM22A-4 treatment (6 months) produced similar ameliorative effects. These results support the hypothesis that targeted activation of TrkB inhibits HD-related degenerative mechanisms, including spine loss, and may provide a disease mechanism-directed therapy for HD and other neurodegenerative conditions.
Tenuifoliside A (TFSA) is a bioactive oligosaccharide ester component of Polygala tenuifolia Wild, a traditional Chinese medicine which was used to manage mental disorders effectively. The neuroprotective and anti-apoptotic effects of TFSA have been demonstrated in our previous studies. The present work was designed to study the molecular mechanism of TFSA on promoting the viability of rat glioma cells C6. We exposed C6 cells to TFSA (or combined with ERK, PI3K and TrkB inhibitors) to examine the effects of TFSA on the cell viability and the expression and phosphorylation of key proteins in the ERK and PI3K signaling pathway. TFSA increased levels of phospho-ERK and phospho-Akt, enhanced release of BDNF, which were blocked by ERK and PI3K inhibitors, respectively (U0126 and LY294002). Moreover, the TFSA caused the enhanced phosphorylation of cyclic AMP response element binding protein (CREB) at Ser133 site, the effect was revoked by U0126, LY294002 and K252a. Furthermore, when C6 cells were pretreated with K252a, a TrkB antagonist, known to significantly inhibit the activity of brain-derived neurotrophic factor (BDNF), blocked the levels of phospho-ERK, phospho-Akt and phosphor-CREB. Taking these results together, we suggested the neuroprotection of TFSA might be mediated through BDNF/TrkB-ERK/PI3K-CREB signaling pathway in C6 glioma cells.
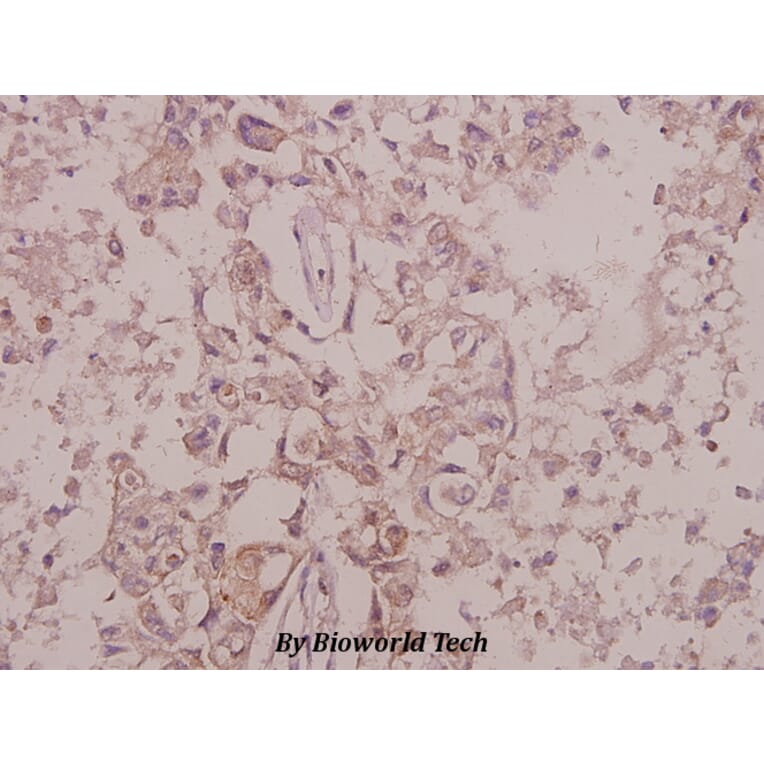
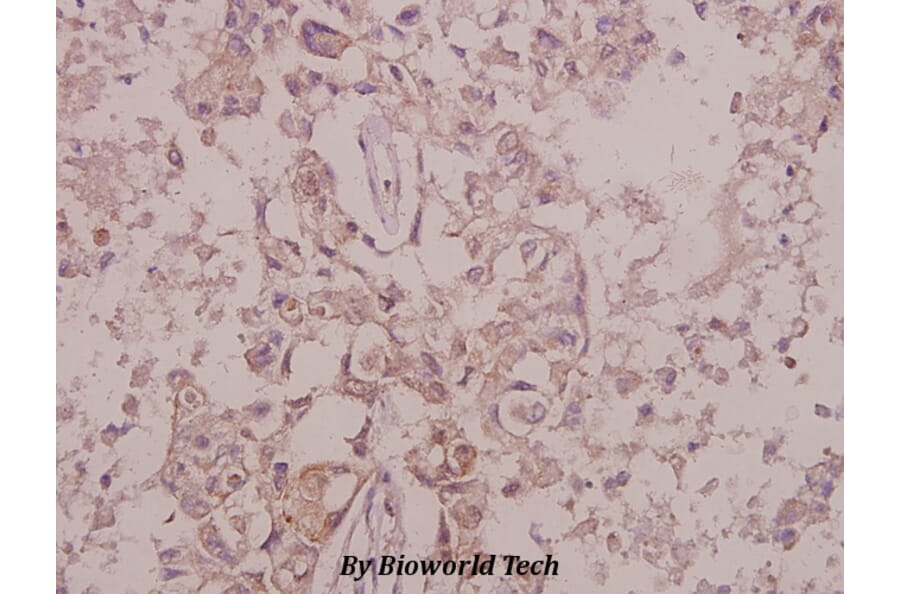



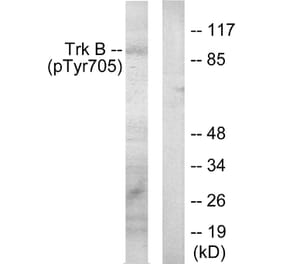
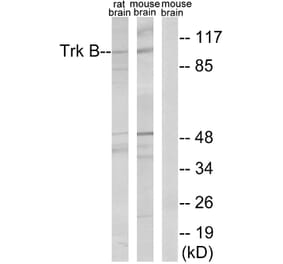

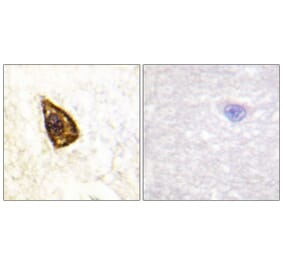
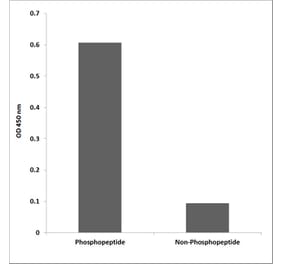
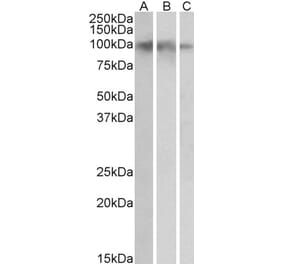
![SDS-PAGE - Anti-TrkB Antibody [Research Grade Biosimilar] - Low endotoxin, Azide free (A324281) - Antibodies.com](https://cdn.antibodies.com/image/catalog/324/A324281_1.jpg?profile=product_alternative)
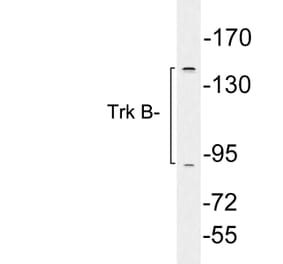
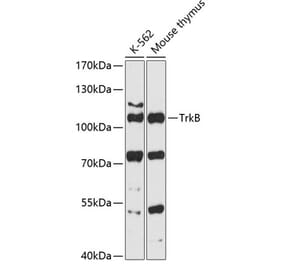
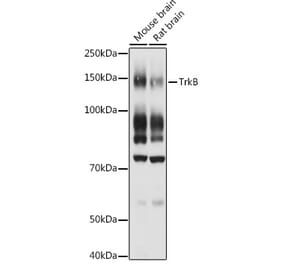
![Western Blot - Anti-TrkB Antibody [ARC53486] (A305512) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305512_1.jpg?profile=product_alternative)