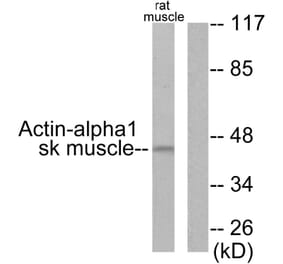
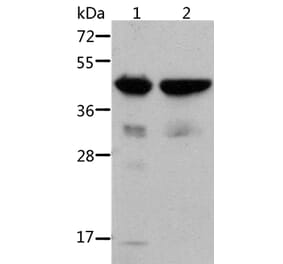
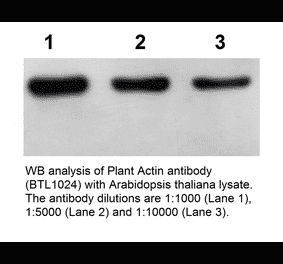
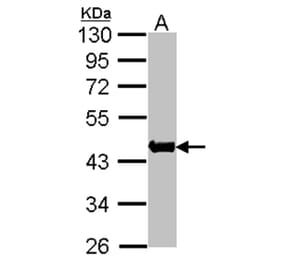
Synonyms
a actin, ACTA, ACTA1, Actin alpha skeletal muscle, actin, alpha 1, skeletal muscle, actin, alpha 1, skeletal muscle 1, Actin, alpha skeletal muscle, actina, actine, ACTS_HUMAN, aktin, Alpha Actin 1, alpha skeletal muscle, Alpha skeletal muscle Actin, alpha-actin, Alpha-actin-1, ASMA, CFTD, CFTD1, CFTDM, MPFD, NEM1, NEM2, NEM3, nemaline myopathy type 3


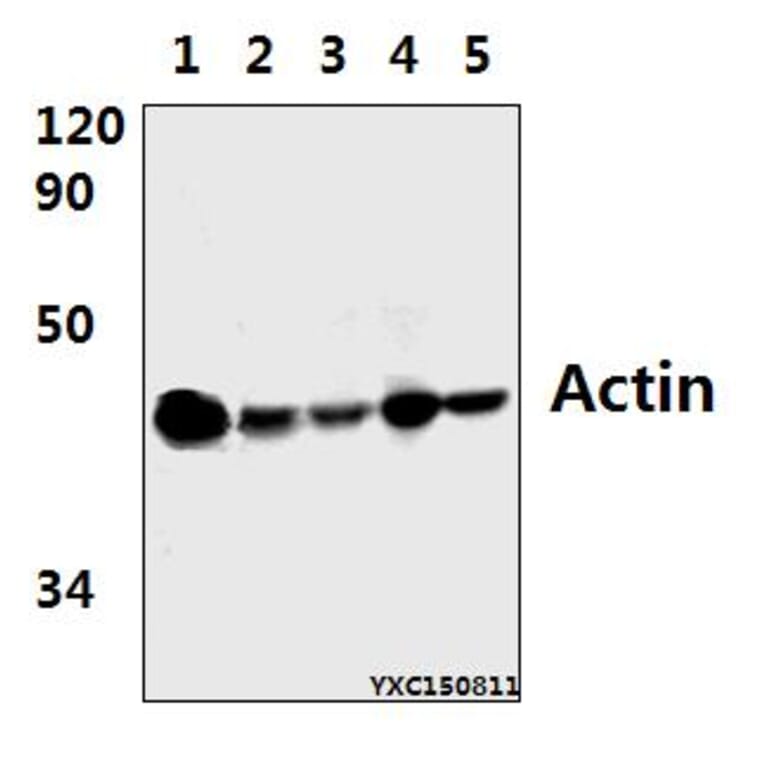


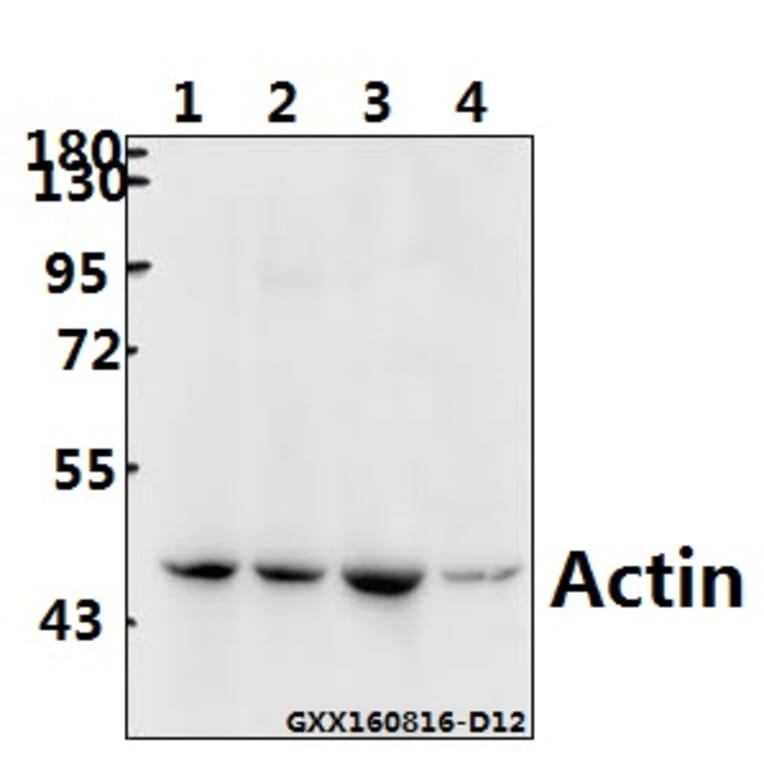


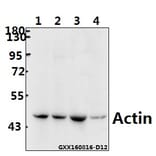
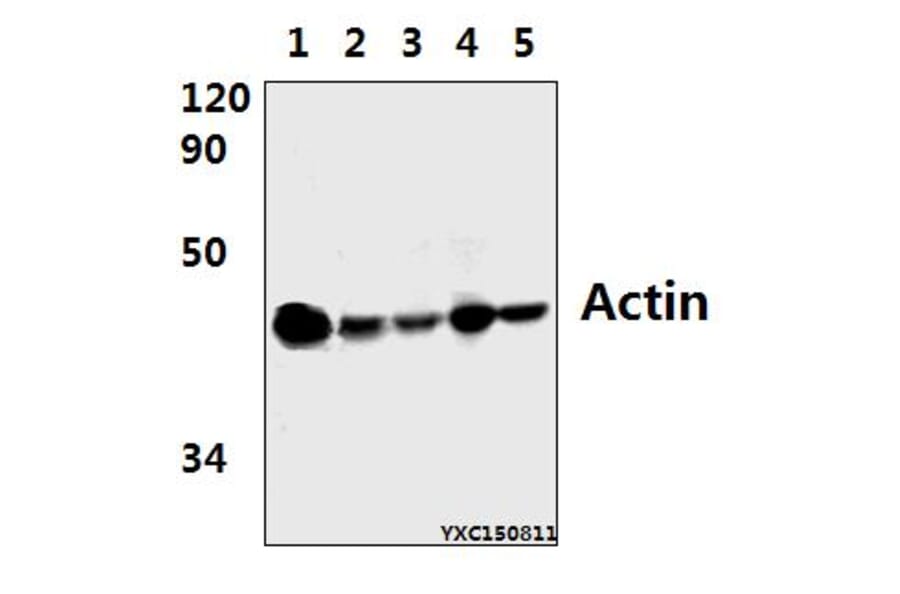
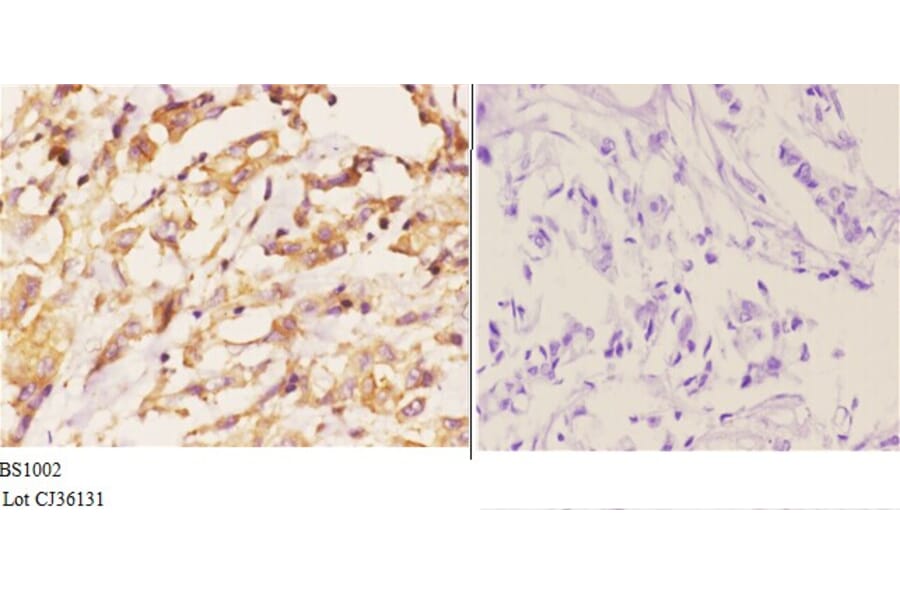


![Immunofluorescence - Anti-Actin Antibody [5J11] (A85388) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85388_1.jpg?profile=product_alternative)