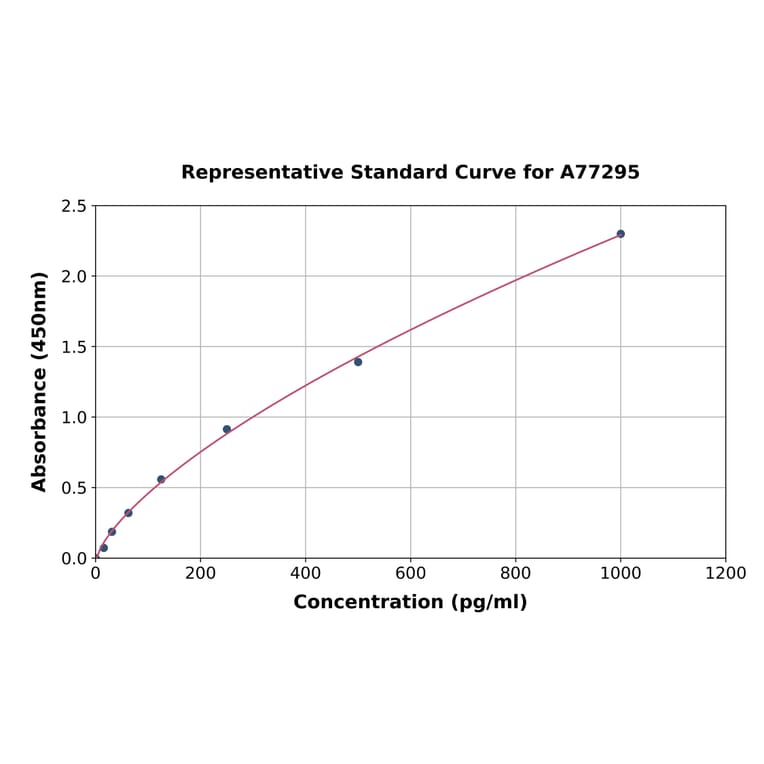
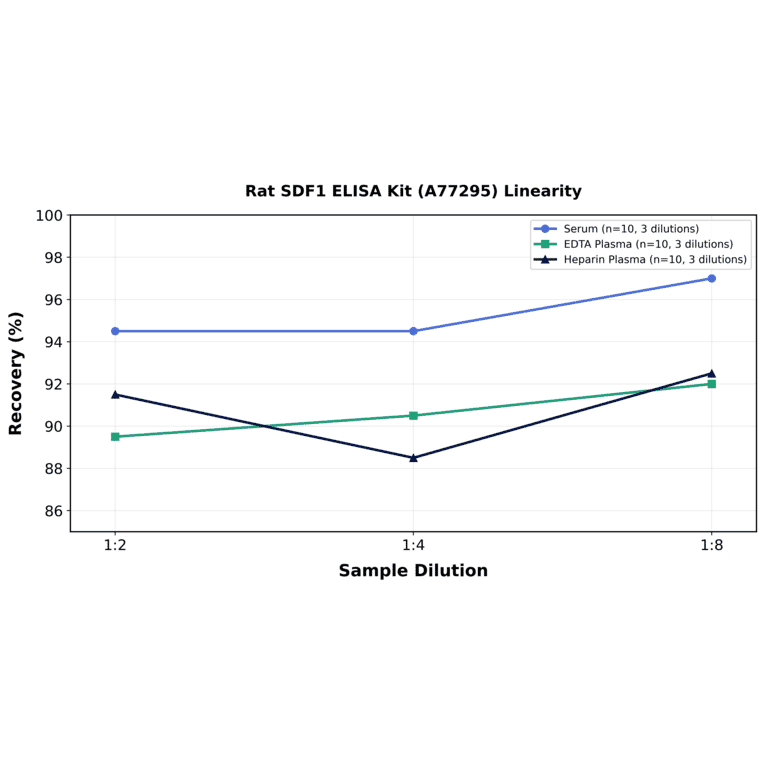
| Sample Type | n | Range | Average |
|---|---|---|---|
| Serum | 10 | 92% - 101% | 93% |
| EDTA Plasma | 10 | 90% - 104% | 96% |
| Heparin Plasma | 10 | 90% - 105% | 93% |
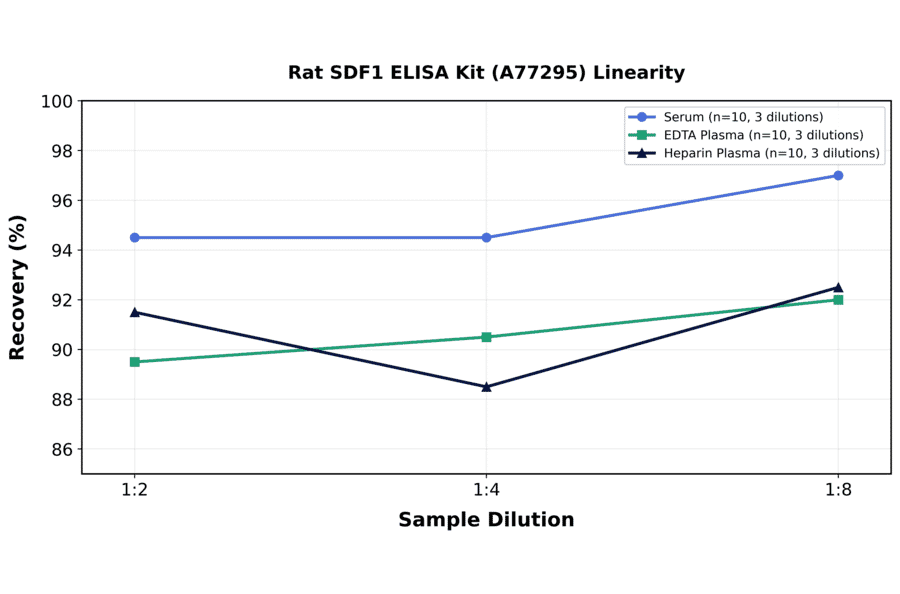
| Sample Type | n | 1:2 | 1:4 | 1:8 |
|---|---|---|---|---|
| Serum | 10 | 89-100% | 85-104% | 89-105% |
| EDTA Plasma | 10 | 83-96% | 82-99% | 85-99% |
| Heparin Plasma | 10 | 83-100% | 81-96% | 85-100% |
| Item | Quantity | Storage |
|---|---|---|
| Pre-Coated 96 Well Microplate | 12 x 8 Well Strips | +4°C |
| Lyopholized Standard | 2 Vials | +4°C |
| Sample Dilution Buffer | 20ml | +4°C |
| Biotinylated Detection Antibody | 120µl | +4°C |
| Antibody Dilution Buffer | 10ml | +4°C |
| HRP-Streptavidin Conjugate | 120µl | +4°C |
| SABC Dilution Buffer | 10ml | +4°C |
| TMB Substrate | 10ml | +4°C |
| Stop Solution | 10ml | +4°C |
| Wash Buffer (25X) | 30ml | +4°C |
| Plate Sealers | 5 Adhesive Strips | - |
| Foil Pouch | 1 Zip-Sealed Pouch | - |
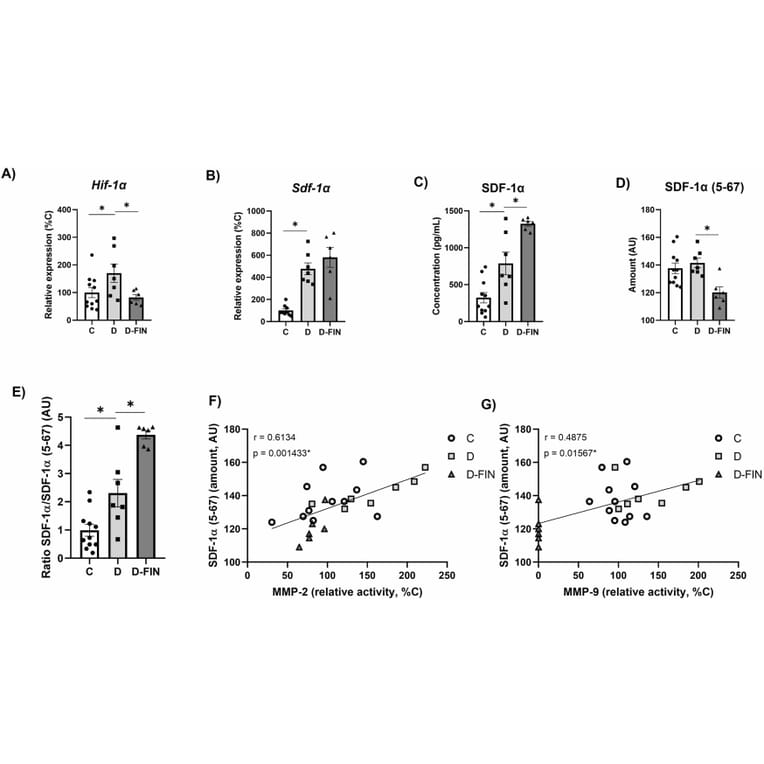
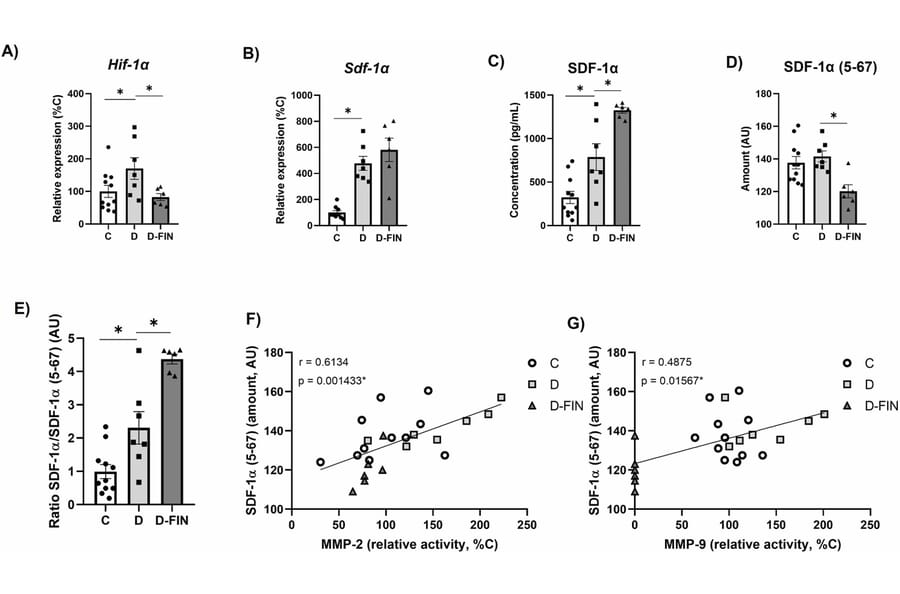
Finerenone (FIN), a non-steroidal mineralocorticoid receptor antagonist, improves kidney and cardiovascular damage in type 1 diabetic (T1DM) Munich Wistar Frömter (MWF) rats with established chronic kidney disease (CKD). We tested whether renal protection involves stromal cell-derived factor 1 (SDF-1)/CXCR4 chemokine axis, a key regulator of tissue repair and stem cell mobilization. T1DM was induced in sixteen-week-old MWF by streptozotocin (15 mg/Kg, i.p.), combined with high fat/high sucrose (HF/HS) diet for 6 weeks (D). A second group (D-FIN) received FIN (10 mg/Kg/day) via the HF/HS diet. Non-diabetic MWF served as controls (C) (n = 11/group). Renal damage was evaluated by histology, RT-qPCR, ELISA, and zymography for matrix metalloproteinase (MMP) activity. Diabetic kidneys in group D showed enhanced glomerulosclerosis, interstitial inflammation, and elevated MMP-2 and MMP-9 activity. FIN treatment significantly reduced these changes, including tubular necrosis and collagen accumulation. Timp-1, Timp-2 and Pai-1 expression remained unchanged across groups. Notably, FIN upregulated SDF-1a and its receptor CXCR4, which are crucial for hematopoietic stem cell (HSC) migration. Conversely, SDF-1a (5-67), a truncated, non-functional form that impairs CXCR4 binding, was reduced with FIN. Immunofluorescence revealed co-localization of CXCR4 with CD34, an HSC marker, in the D-FIN group. We conclude that FIN mitigates diabetic kidney injury in MWF rats by promoting HSC (CD34?) recruitment to the kidney. This is mediated through decreased MMP-2/9 activity, upregulation of the SDF-1a/CXCR4 axis, and reduced expression of the non-functional SDF-1a (5-67) form. These findings support a novel mechanism of FIN-induced renal protection involving stem cell mobilization.
During Type 2 diabetes (T2D), the immune system becomes dysregulated, adopting a pro-inflammatory phenotype and secreting cytokines that exacerbate inflammation. Human amylin (i.e., islet amyloid polypeptide) secreted from the pancreas of HIP rats during T2D has been shown to deposit in the microvasculature and lead to neurodegeneration and cognitive decline. Although T2D is linked to neuroinflammation and cognitive impairment, whether and how peripheral immune changes, driven in part by amyloidogenic amylin deposition, shape these outcomes is not well understood. We investigated how the accumulation of pancreatic human amylin impacts the peripheral and neuroimmune response in transgenic rat and mouse models with endogenous overexpression. In rats, hypersecretion of pancreatic human amylin reduced adaptive and innate immune cells in the spleen and brain, the latter associated with the downregulation of cerebrovascular ICAM-1, which is responsible for immune cell migration into the brain. Acute systemic injections of aggregated human amylin in wild-type (WT) rats led to immune alterations in the blood only, suggesting brain-specific changes may require chronic exposure. In mice that hypersecrete pancreatic human amylin, we identified defects in B cell development, including reduced B cell populations in the spleen and bone marrow. This immune dysregulation was associated with upregulated CXCR4 expression in precursor B cells, suggesting a potential mechanism for systemic B cell losses during chronic T2D. To further support these findings in human disease, single-cell sequencing of patients with T2D revealed elevated CXCR4 expression across multiple B cell subsets, highlighting the relevance of this axis in diabetic immune dysregulation. Taken together, our findings indicate that T2D-associated amylin may contribute to T2D by altering both peripheral and neuroimmune responses. Moreover, the CXCL12/CXCR4 signaling axis emerges as a promising target for immunotherapeutic strategies to mitigate type-2 diabetic cognitive decline and neurodegeneration.