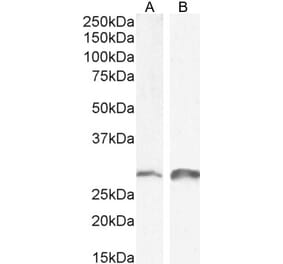
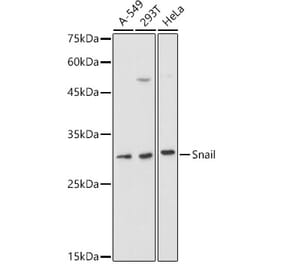
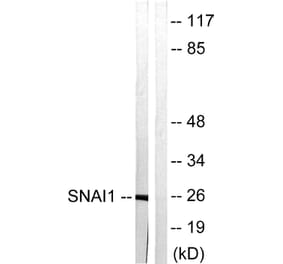
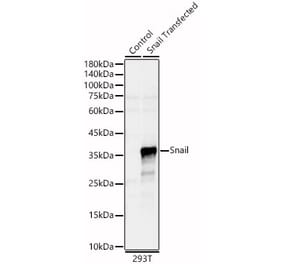
Unconjugated
Glycogen storage disease type Ib (GSD-Ib), caused by a deficiency of the glucose-6-phosphate transporter (G6PT), is characterized by impaired glucose homeostasis and progressive renal dysfunction. Using G6pt-deficient (G6pt-/-) mice, which closely recapitulate the pathophysiology of GSD-Ib, we previously demonstrated that GSD-Ib nephropathy is marked by disrupted renal homeostasis, acute kidney injury, and fibrosis. However, due to the severity of the metabolic defect, G6pt-/- mice typically fail to survive beyond three weeks of age, limiting the study of disease progression in adulthood. To overcome this limitation, we generated liver human G6PT-augmented, kidney G6PT-deficient (L-hG6PT/K-/-) mice, which restore hepatic G6PT expression to support survival while maintaining G6PT deficiency in the kidney. We show that adult L-hG6PT/K-/- mice develop renal abnormalities similar to those seen in 3-week-old global G6pt-/- mice. Importantly, unlike the younger cohort, 12-week-old L-hG6PT/K-/- mice exhibit a progressive decline in renal function accompanied by marked fibrosis. These findings establish L-hG6PT/K-/- mice as a robust and physiologically relevant model for investigating the mechanisms and progression of GSD-Ib nephropathy in mature animals.
Glycogen storage disease type Ib (GSD-Ib) results from a deficiency in the ubiquitously expressed glucose-6-phosphate transporter (G6PT), which partners with either the liver/kidney-specific glucose-6-phosphatase-a (G6Pase-a, G6PC1) or the ubiquitously expressed G6Pase-ß (G6PC3) to produce glucose from G6P. A deficiency in G6Pase-a causes GSD-Ia. Since G6Pase-a is more active than G6Pase-ß, glucose homeostasis is mainly maintained by the G6PT/G6Pase-a complex, and both GSD-Ia and GSD-Ib share metabolic defects and renal disease. GSD-Ia nephropathy is characterized by glomerulosclerosis and fibrosis, partly driven by Wnt/ß-catenin signaling. In this study, we show that G6pt-/- (GSD-Ib) mice exhibit similar features, including Wnt/ß-catenin-mediated fibrosis, but with significantly higher renal triglyceride levels compared to age-matched GSD-Ia mice during weeks 1 to 3 postnatal development, leading to an early onset of more severe kidney disease. G6Pase-ß is highly expressed in the kidney but minimally in the liver, distinguishing the GSD-I subtypes. In GSD-Ia, G6PT/G6Pase-a activity is absent in both the liver and kidney, while G6PT/G6Pase-ß is functionally active in the kidney. In GSD-Ib, both G6PT complexes are absent in the kidney and liver. Our results suggest that the less active G6PT/G6Pase-ß complex plays a protective role in GSD-Ia kidney.