

Unconjugated
Mutations in genes involved in glycosylphosphatidylinositol (GPI) anchor biosynthesis underlie a group of congenital syndromes characterized by severe neurodevelopmental defects. GPI anchored proteins have diverse roles in cell adhesion, signaling, metabolism and complement regulation. Over 30 enzymes are required for GPI anchor biosynthesis and PIGA is involved in the first step of this process. A hypomorphic mutation in the X-linked PIGA gene (c.1234C>T) causes multiple congenital anomalies hypotonia seizure syndrome 2 (MCAHS2), indicating that even partial reduction of GPI anchored proteins dramatically impairs central nervous system development, but the mechanism is unclear. Here, we established a human induced pluripotent stem cell (hiPSC) model containing the PIGAc.1234C>T mutation to study the effects of a hypomorphic allele of PIGA on neuronal development. Neuronal differentiation from neural progenitor cells generated by EB formation in PIGAc.1234C>T is significantly impaired with decreased proliferation, aberrant synapse formation and abnormal membrane depolarization. The results provide direct evidence for a critical role of GPI anchor proteins in early neurodevelopment. Furthermore, neural progenitors derived from PIGAc.1234C>T hiPSCs demonstrate increased susceptibility to complement-mediated cytotoxicity, suggesting that defective complement regulation may contribute to neurodevelopmental disorders.
Congenital and acquired deficiencies of complement regulatory proteins are associated with pathologic complement activation in several renal diseases. To elucidate the mechanisms by which renal tubular epithelial cells (TECs) control the complement system, we examined the expression of complement regulatory proteins by the cells. We found that Crry is the only membrane-bound complement regulator expressed by murine TECs, and its expression is concentrated on the basolateral surface. Consistent with the polarized localization of Crry, less complement activation was observed when the basolateral surface of TECs was exposed to serum than when the apical surface was exposed. Furthermore, greater complement activation occurred when the basolateral surface of TECs from Crry(-/-)fB(-/-) mice was exposed to normal serum compared with TECs from wild-type mice. Complement activation on the apical and basolateral surfaces was also greater when factor H, an alternative pathway regulatory protein found in serum, was blocked from interacting with the cells. Finally, we injected Crry(-/-)fB(-/-) and Crry(+/+)fB(-/-) mice with purified factor B (an essential protein of the alternative pathway). Spontaneous complement activation was seen on the tubules of Crry(-/-)fB(-/-) mice after injection with factor B, and the mice developed acute tubular injury. These studies indicate that factor H and Crry regulate complement activation on the basolateral surface of TECs and that factor H regulates complement activation on the apical surface. However, congenital deficiency of Crry or reduced expression of the protein on the basolateral surface of injured cells permits spontaneous complement activation and tubular injury.
![Western Blot - Anti-CD59 Antibody [MEM-43/5] (A85694) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85695_162.jpg?profile=product_top)
![Western Blot - Anti-CD59 Antibody [MEM-43/5] (A85694) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85695_162.jpg?profile=product_top_thumb)
![Western Blot - Anti-CD59 Antibody [MEM-43/5] (A85694) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85695_162.jpg?profile=product_image)












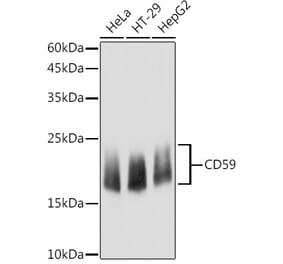
![Western Blot - Anti-CD59 Antibody [ARC0896] (A305532) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305532_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-CD59 Antibody [MACIF/629] (A250740) - Antibodies.com](https://cdn.antibodies.com/image/catalog/250/A250740_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-CD59 Antibody [MACIF/629] - BSA and Azide free (A253920) - Antibodies.com](https://cdn.antibodies.com/image/catalog/253/A253920_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-CD59 Antibody [193-27] (A251079) - Antibodies.com](https://cdn.antibodies.com/image/catalog/251/A251079_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-CD59 Antibody [193-27] - BSA and Azide free (A254356) - Antibodies.com](https://cdn.antibodies.com/image/catalog/254/A254356_1.jpg?profile=product_alternative)