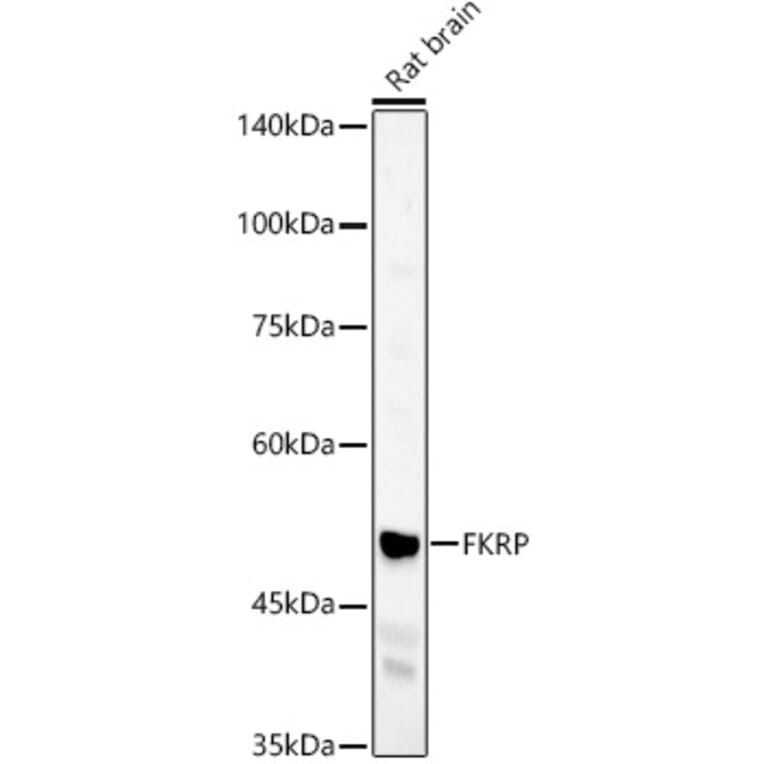
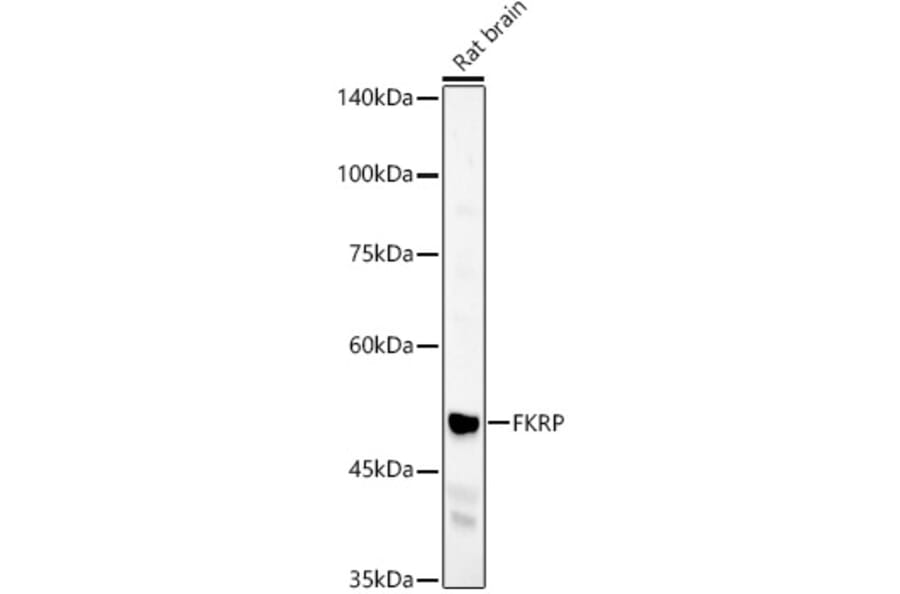
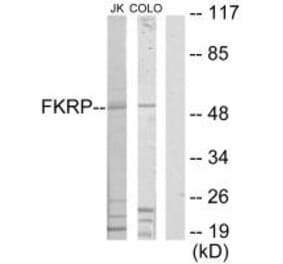
Western blot analysis of Rat brain, using Anti-FKRP Antibody (A8549) at 1:400 dilution. The secondary antibody was Goat Anti-Rabbit IgG H&L Antibody (HRP) at 1:10,000 dilution. Lysates/proteins were present at 25µg per lane. The blocking buffer used was 3% non-fat dry milk in TBST. Detection was with a ECL Basic Kit. Exposure time: 60s.
Publishing research using Anti-FKRP Antibody (A8549)? Please let us know so that we can list the citation on this page.
Alternative products to Anti-FKRP Antibody (A8549)