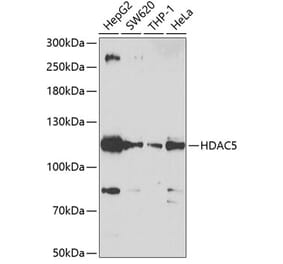
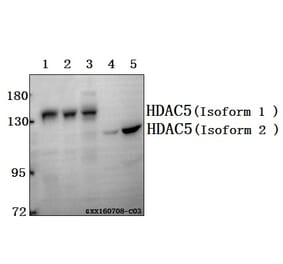
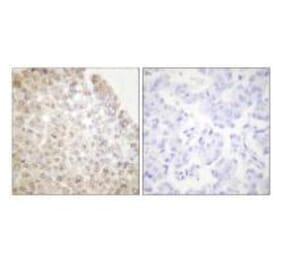
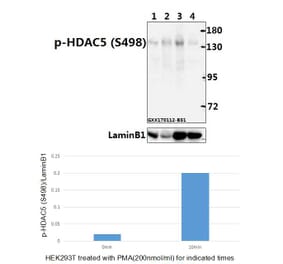
Unconjugated
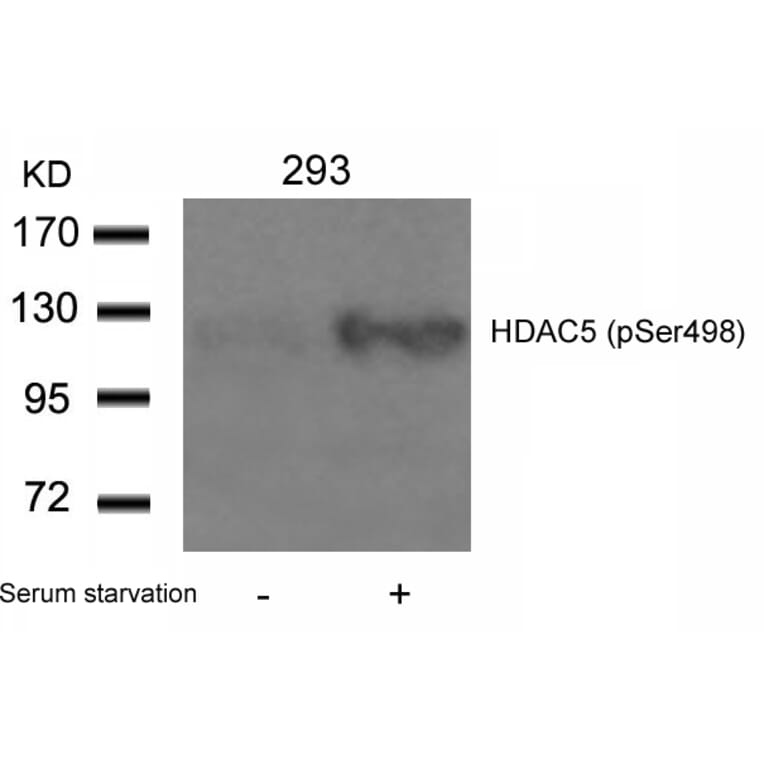
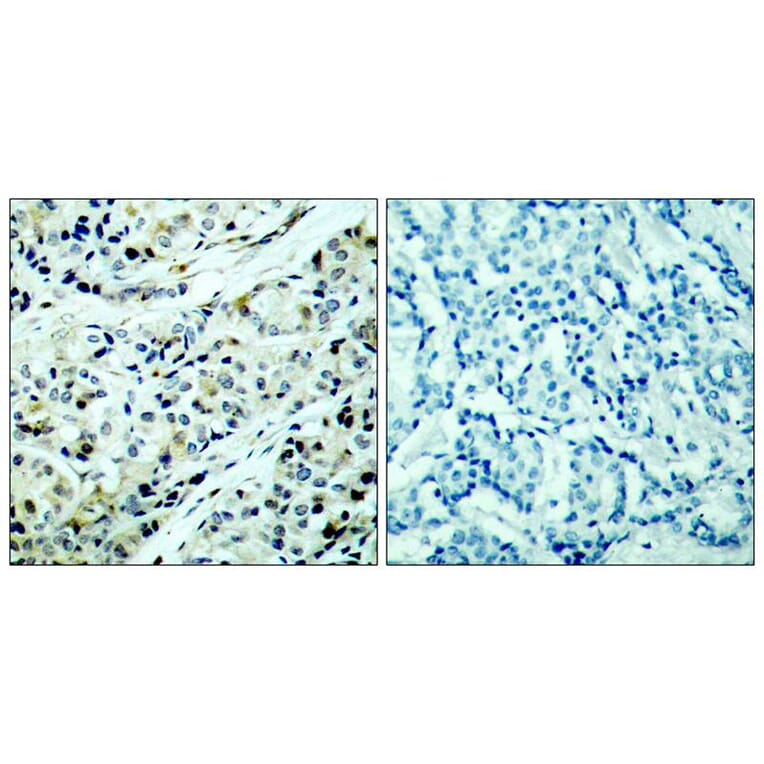
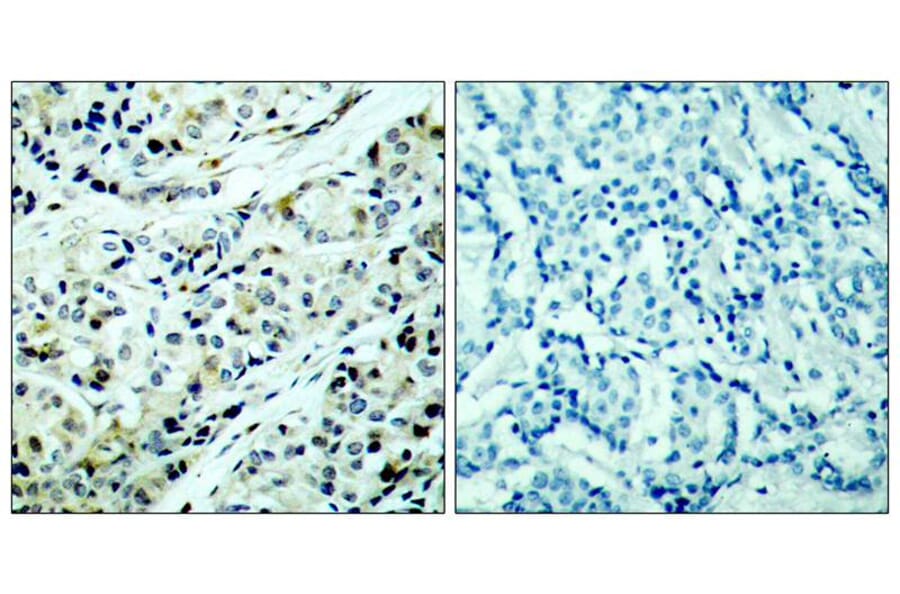
Polycystin 1 and polycystin 2 are large transmembrane proteins, which, when mutated, cause autosomal dominant polycystic kidney disease (ADPKD), a highly prevalent human genetic disease. The polycystins are thought to form a receptor-calcium channel complex in the plasma membrane of renal epithelial cells and elicit a calcium influx in response to mechanical stimulation, such as fluid flow across the apical surface of renal epithelial cells. The functional role of the polycystins in mechanosensation remains largely unknown. Here, we found that myocyte enhancer factor 2C (MEF2C) and histone deacetylase 5 (HDAC5), two key regulators of cardiac hypertrophy, are targets of polycystin-dependent fluid stress sensing in renal epithelial cells in mice. We show that fluid flow stimulation of polarized epithelial monolayers induced phosphorylation and nuclear export of HDAC5, which are crucial events in the activation of MEF2C-based transcription. Kidney-specific knockout of Mef2c, or genetrap-inactivation of a MEF2C transcriptional target, MIM, resulted in extensive renal tubule dilation and cysts, whereas Hdac5 heterozygosity or treatment with TSA, an HDAC inhibitor, reduced cyst formation in Pkd2(-/-) mouse embryos. These findings suggest a common signaling motif between myocardial hypertrophy and maintenance of renal epithelial architecture, and a potential therapeutic approach to treat ADPKD.
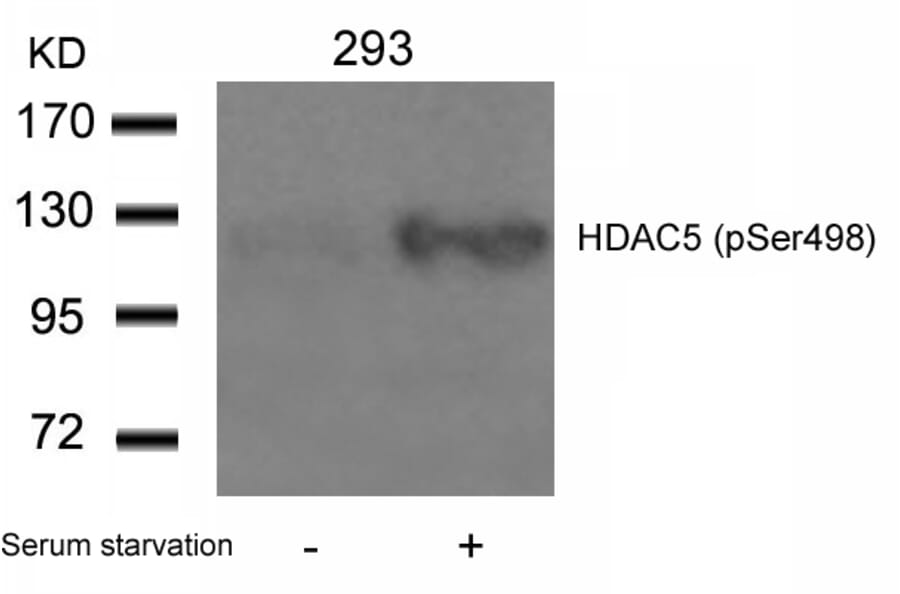
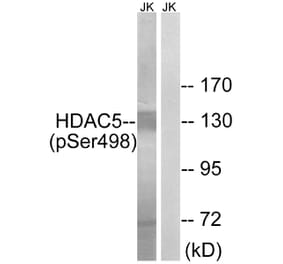
Fluid shear stress generated by steady laminar blood flow protects vessels from atherosclerosis. Krüppel-like factor 2 (KLF2) and endothelial nitric oxide synthase (eNOS) are fluid shear stress-responsive genes and key mediators in flow anti-inflammatory and antiatherosclerotic actions. However, the molecular mechanisms underlying flow induction of KLF2 and eNOS remain largely unknown. Here, we show a novel role of histone deacetylase 5 (HDAC5) in flow-mediated KLF2 and eNOS expression. We found for the first time that fluid shear stress stimulated HDAC5 phosphorylation and nuclear export in endothelial cells through a calcium/calmodulin-dependent pathway. Consequently, flow induced the dissociation of HDAC5 and myocyte enhancer factor-2 (MEF2) and enhanced MEF2 transcriptional activity, which leads to expression of KLF2 and eNOS. Adenoviral overexpression of a HDAC5 phosphorylation-defective mutant (Ser259/Ser498 were replaced by Ala259/Ala498, HDAC5-S/A), which shows resistance to flow-induced nuclear export, suppressed flow-mediated MEF2 transcriptional activity and expression of KLF2 and eNOS. Importantly, HDAC5-S/A attenuated the flow-inhibitory effect on monocyte adhesion to endothelial cells. Taken together, our results reveal that phosphorylation-dependent derepression of HDAC5 mediates flow-induced KLF2 and eNOS expression as well as flow anti-inflammation, and suggest that HDAC5 could be a potential therapeutic target for the prevention of atherosclerosis.