Unconjugated
Chronic inflammation-promoted metastasis has been considered as a major challenge in cancer therapy. Pro-inflammatory cytokine TNFα can induce cancer invasion and metastasis associated with epithelial-mesenchymal transition (EMT). However, the underlying mechanisms are not entirely clear. In this study, we showed that TNFα induces EMT in human HCT116 cells and thereby promotes colorectal cancer (CRC) invasion and metastasis. TNFα-induced EMT was characterized by acquiring mesenchymal spindle-like morphology and increasing the expression of N-cadherin and fibronectin with a concomitant decrease of E-cadherin and Zona occludin-1(ZO-1). TNFα treatment also increased the expression of transcription factor Snail, but not Slug, ZEB1 and Twist. Overexpression of Snail induced a switch from E-cadherin to N-cadherin expression in HCT116 cells, which is a characteristic of EMT. Conversely, knockdown of Snail significantly attenuated TNFα-induced EMT in HCT116 cells, suggesting that Snail plays a crucial role in TNFα-induced EMT. Interestingly, exposure to TNFα rapidly increased Snail protein expression and Snail nuclear localization but not mRNA level upregulation. Finally, we demonstrated that TNFα elevated Snail stability by activating AKT pathway and subsequently repressing GSK-3β activity and decreasing the association of Snail with GSK-3β. Knockdown of GSK-3β further verified our finding. Taken together, these results revealed that AKT/GSK-3β-mediated stabilization of Snail is required for TNFα-induced EMT in CRC cells. Our study provides a better understanding of inflammation-induced CRC metastasis.
The IκB kinase (IKK)/NF-κB pathway has been shown to be a major regulator in cell survival. However, the mechanisms through which IKK mediates cell death are not clear. In this study, we showed that IKK-β contributed to hydrogen peroxide (H(2)O(2))-induced cell death independent of the NF-κB pathway. Our results demonstrated that the pro-death function of IKK-β under oxidative stress was mediated by p85 S6K1 (S6 kinase 1), but not p70 S6K1 through a rapamycin-insensitive and mammalian target of rapamycin complex 1 kinase-independent mechanism. We found that IKK-β associated with p85, but not p70 S6K1, which was required for H(2)O(2)-induced activation of p85 S6K1. IKK-β and p85 S6K1 contributed to H(2)O(2)-induced phosphorylation of Mdm2 (S166) and p53 accumulation. p85 S6K1 is critical for IKK-β-mediated cell death. Thus, these findings established a novel oxidative stress-responsive pathway that involves IKK-β, p85 S6K1 and Mdm2, which is response for H(2)O(2)-induced cell death. Our results have important implications for IKK-β and p85 S6K1 as potential targets for the prevention of diseases involved in oxidative stress-induced aberrant cell death.

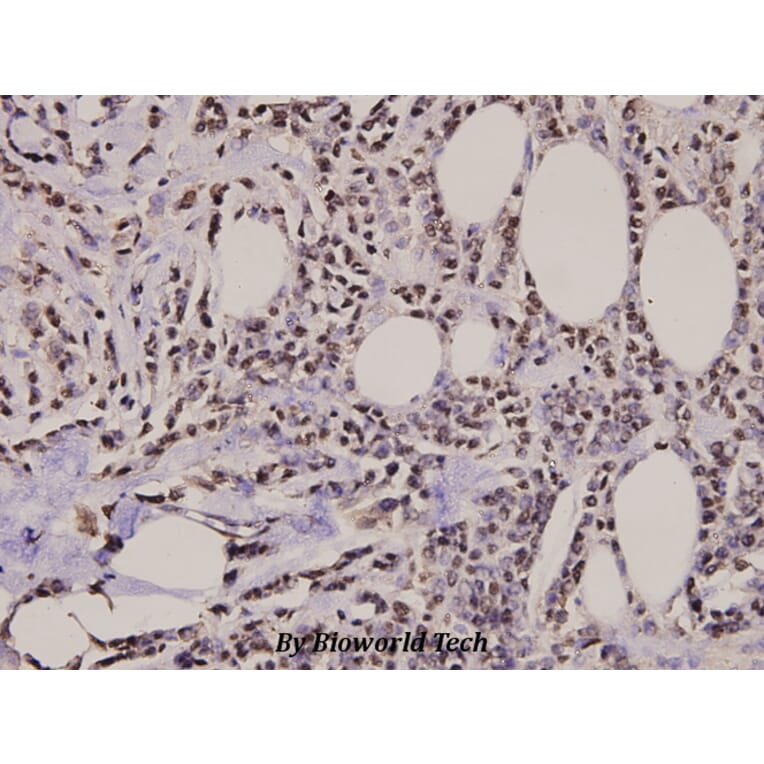
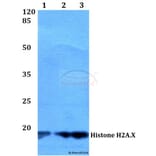
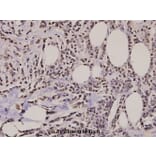


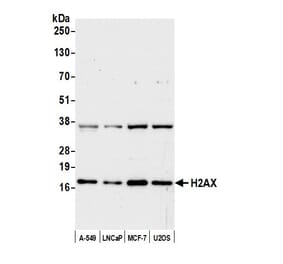

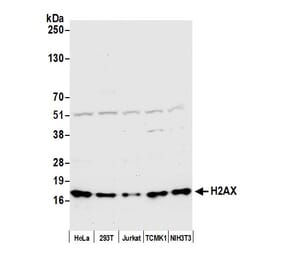
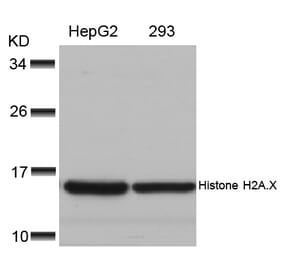
![Western Blot - Anti-Histone H2A.X (phospho Ser139) Antibody [ARC0110] (A308961) - Antibodies.com](https://cdn.antibodies.com/image/catalog/308/A308961_1.jpg?profile=product_alternative)
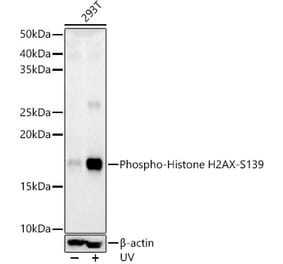
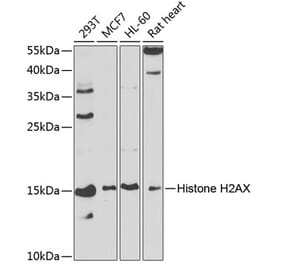
![Western Blot - Anti-Histone H2AX Antibody [RM214] (A121318) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121318_1.png?profile=product_alternative)