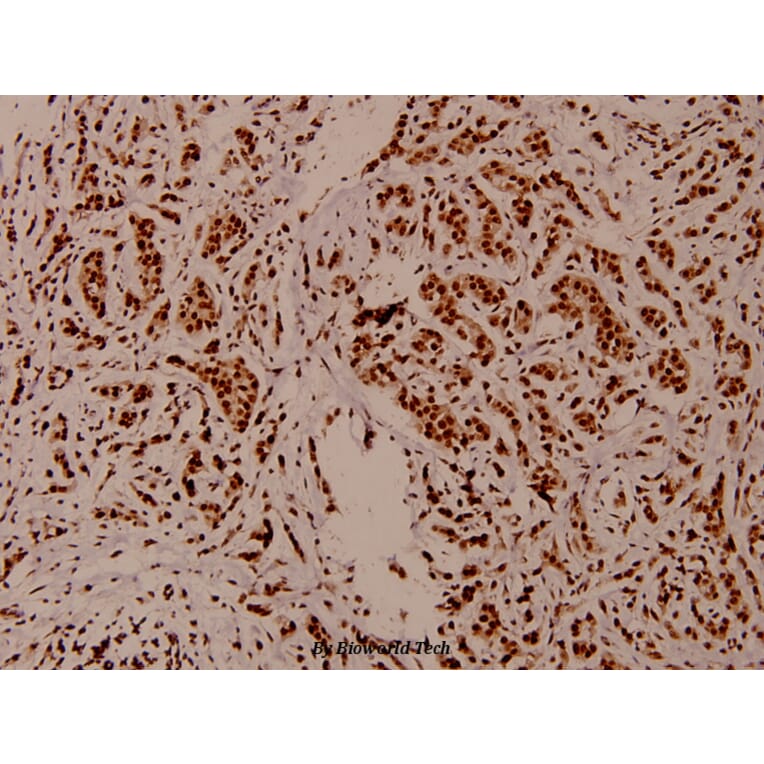
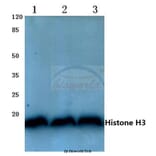
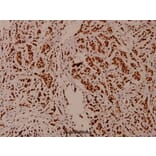
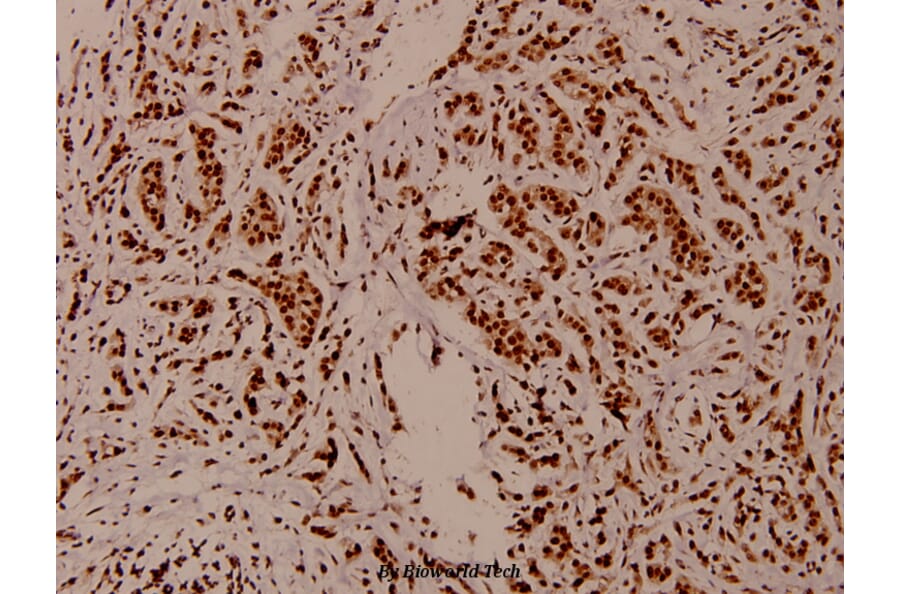
Synonyms
H3-3A, H3-5, H3-clustered histone 13, H3-clustered histone 14, H3-clustered histone 15, H3.3A, H3C1, H3C15, H3F3, H3F3A, H3F3C, H3FA, HIST1H3A, HIST2H3A, Histone H3.1, Histone H3.2, Histone H3.3, Histone H3.3C, Histone H3.5, Histone H3/a, Histone H3/b, Histone H3/c, Histone H3/d, Histone H3/f, Histone H3/m, Histone H3/o