

Unconjugated
The receptor for advanced-glycation end products (RAGE) is upregulated in various cancers and has been associated with tumor progression, but little is known about its expression and regulation by microRNAs (miRNAs) in esophageal squamous cell carcinoma (ESCC). Here, we describe miR-185, which represses RAGE expression, and investigate the biological role of miR-185 in ESCC. In this study, we found that the high level of RAGE expression in 29 pairs of paraffin-embedded ESCC tissues was correlated positively with the depth of invasion by immunohistochemistry, suggesting that RAGE was involved in ESCC. We used bioinformatics searches and luciferase reporter assays to investigate the prediction that RAGE was regulated directly by miR-185. Besides, overexpression of miR-185 in ESCC cells was accompanied by 27% (TE-11) and 49% (Eca-109) reduced RAGE expression. The effect was further confirmed in RAGE protein by immunofluorescence in both cell lines. The effects were reversed following cotransfection with miR-185 and high-level expression of the RAGE vector. Furthermore, the biological role of miR-185 in ESCC cell lines was investigated using assays of cell viability, Ki-67 staining, and cell migration and invasion, as well as in a xenograft model. We found that overexpression of miR-185 inhibited migration and invasion by ESCC cells in vitro and reduced their capacity to develop distal pulmonary metastases in vivo partly through the RAGE/heat shock protein 27 pathway. Interestingly, in clinical specimens, the level of plasma miR-185 expression was decreased significantly (P = 0.002) in patients with ESCC [0.500; 95% confidence interval (CI) 0.248-1.676] compared with healthy controls (2.410; 95% CI 0.612-5.671). The value of the area under the receiver-operating characteristic curve was 0.73 (95% CI 0.604-0.855). In conclusion, our findings shed novel light on the role of miR-185/RAGE in ESCC metastasis, and plasma miR-185 has potential as a novel diagnostic biomarker in ESCC.
The crosstalk of intracellular signaling pathways is extremely complex. Previous studies have shown that there is a potential crosstalk between MAPKs and NF-κB signaling pathways. It has been reported that JNK regulates cell survival under some conditions. But the molecular mechanism through which JNK regulates cell survival is still unclear. In the present study, we hypothesized that there was a crosstalk between JNK and NF-κB signaling pathway regulating cell survival and HSP27 phosphorylation mediates such a crosstalk. Our data showed that in HepG2 cells, suppression of JNK activation by a specific inhibitor or overexpression of JNK inactive mutant enhanced TNF-α-induced apoptosis. In addition, reduction of JNK activation attenuated HSP27 phosphorylation envoked by TNF-α, especially the phosphorylation of HSP27 at serine 78 residue. Our results also showed that suppression of JNK activation reduced the degradation of IκB-α, but did not affect IKK phosphorylation upon TNF-α stimulation. Co-immunoprecipitation experiments demonstrated that JNK regulated the degradation of IκB-α through promoting the formation of HSP27/IKK/IκB-α ternary complex in response to TNF-α. Suppression of JNK activation hindered HSP27 phosphorylation at Ser78 residue and subsequently reduced the interaction between IKK and IκB-α. Taken together, our study suggests that through modulation the phosphorylation of HSP27, JNK plays an important roles in cell survival via regulating NF-κB signaling pathway.




![Immunohistochemistry - Anti-HSP27 Antibody [G3.1] (A248872) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248872_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-HSP27 Antibody [8A7] (A304709) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304709_1.png?profile=product_alternative)
![Immunohistochemistry - Anti-HSP27 Antibody [G3.1] - BSA and Azide free (A252052) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252052_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-HSP27 Antibody [SPM252] (A248873) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248873_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-HSP27 Antibody [SPM252] - BSA and Azide free (A252053) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252053_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-HSP27 Antibody [HSPB1/774] (A248874) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248874_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-HSP27 Antibody [HSPB1/774] - BSA and Azide free (A252054) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252054_1.jpg?profile=product_alternative)
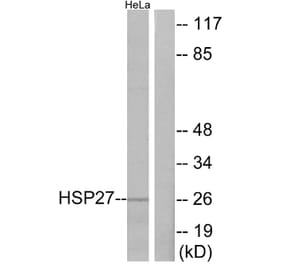
![SDS-PAGE - Anti-HSP27 Antibody [CPTC-HSPB1-2] - BSA and Azide free (A252055) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252055_1.jpg?profile=product_alternative)
![SDS-PAGE - Anti-HSP27 Antibody [CPTC-HSPB1-2] (A248875) - Antibodies.com](https://cdn.antibodies.com/image/catalog/248/A248875_1.jpg?profile=product_alternative)
![Western Blot - Anti-HSP27 Antibody [5D12-A12] (A304734) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304734_1.png?profile=product_alternative)