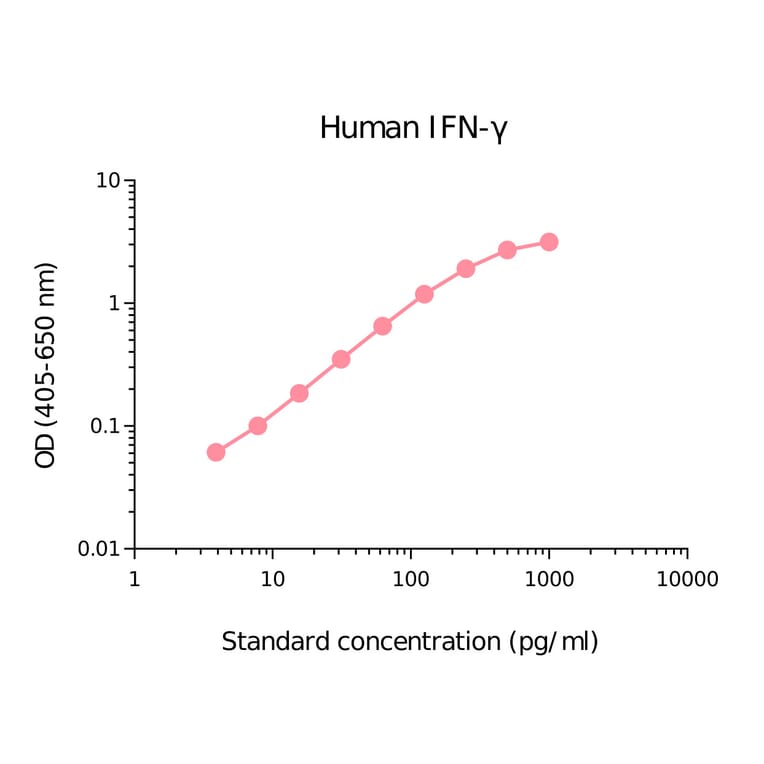
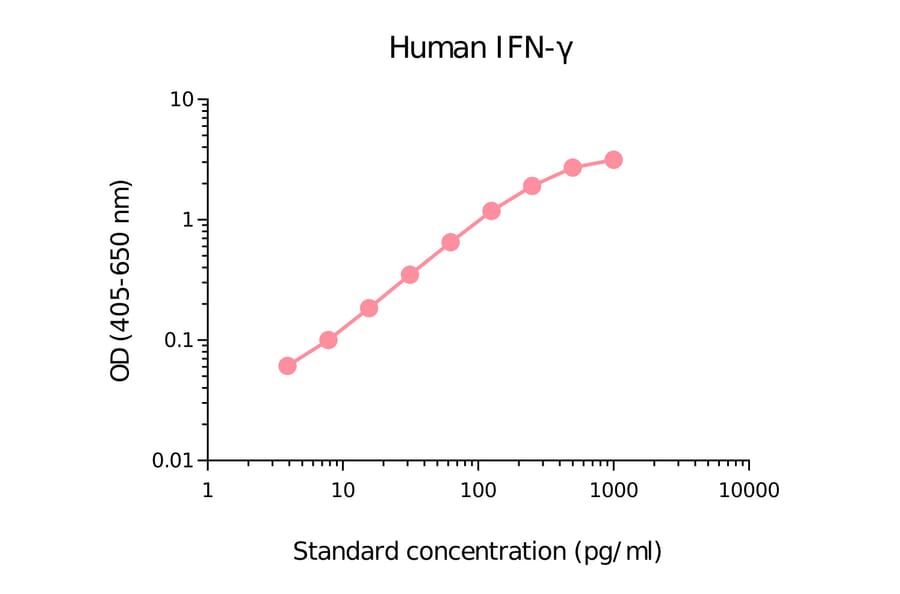
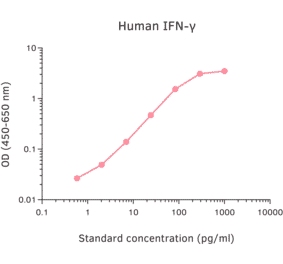
Epstein-Barr virus (EBV) is a tumor virus that establishes lifelong infection in most of humanity, despite eliciting strong and stable virus-specific immune responses. EBV encodes at least 44 miRNAs, most of them with unknown function. Here, we show that multiple EBV miRNAs modulate immune recognition of recently infected primary B cells, EBV's natural target cells. EBV miRNAs collectively and specifically suppress release of proinflammatory cytokines such as IL-12, repress differentiation of naive CD4(+) T cells to Th1 cells, interfere with peptide processing and presentation on HLA class II, and thus reduce activation of cytotoxic EBV-specific CD4(+) effector T cells and killing of infected B cells. Our findings identify a previously unknown viral strategy of immune evasion. By rapidly expressing multiple miRNAs, which are themselves nonimmunogenic, EBV counteracts recognition by CD4(+) T cells and establishes a program of reduced immunogenicity in recently infected B cells, allowing the virus to express viral proteins required for establishment of life-long infection.
Psoriasis vulgaris is a common T cell-mediated inflammatory skin disease with a suspected autoimmune pathogenesis. The human leukocyte antigen (HLA) class I allele, HLA-C*06:02, is the main psoriasis risk gene. Epidermal CD8(+) T cells are essential for psoriasis development. Functional implications of HLA-C*06:02 and mechanisms of lesional T cell activation in psoriasis, however, remained elusive. Here we identify melanocytes as skin-specific target cells of an HLA-C*06:02-restricted psoriatic T cell response. We found that a Va3S1/Vß13S1 T cell receptor (TCR), which we had reconstituted from an epidermal CD8(+) T cell clone of an HLA-C*06:02-positive psoriasis patient specifically recognizes HLA-C*06:02-positive melanocytes. Through peptide library screening, we identified ADAMTS-like protein 5 (ADAMTSL5) as an HLA-C*06:02-presented melanocytic autoantigen of the Va3S1/Vß13S1 TCR. Consistent with the Va3S1/Vß13S1-TCR reactivity, we observed numerous CD8(+) T cells in psoriasis lesions attacking melanocytes, the only epidermal cells expressing ADAMTSL5. Furthermore, ADAMTSL5 stimulation induced the psoriasis signature cytokine, IL-17A, in CD8(+) T cells from psoriasis patients only, supporting a role as psoriatic autoantigen. This unbiased analysis of a TCR obtained directly from tissue-infiltrating CD8(+) T cells reveals that in psoriasis HLA-C*06:02 directs an autoimmune response against melanocytes through autoantigen presentation. We propose that HLA-C*06:02 may predispose to psoriasis via this newly identified autoimmune pathway.