Unconjugated
The mammalian target of rapamycin (mTOR) pathway is a crucial cellular signaling hub, which integrates internal and external cues to modulate the cell cycle, protein synthesis and metabolism. The present study hypothesized that inhibiting mTOR signaling may induce cells to enter lower and more stable bioenergetic states, in which neurons have greater resistance to various insults. Neurotrophin withdrawal from photoreceptor cells (661W cells) was mimicked using serum deprivation, and the neuroprotective mechanisms were studied following suppression of the mTOR pathway. Treatment with an mTOR specific inhibitor, rapamycin, reduced intracellular levels of reactive oxygen species, suppressed oxidative stress, and attenuated mitochondrial dysfunction. In addition, inhibiting mTOR signaling induced a G2/M cell cycle arrest, thus providing an opportunity to repair damaged DNA and block the cell death cascade. These results suggested that inhibition of mTOR had a neuroprotective effect on serum‑deprived 661W cells. In conclusion, the mTOR pathway is a critical molecular signal for cell cycle regulation and energy metabolism, and inhibiting the mTOR pathway may attenuate neurotrophin withdrawal‑induced damage. These observations may provide evidence for the treatment of retinal degenerative disease, since inducing neurons into a lower and more stable bioenergetic state by blocking mTOR signaling may slow the progression of neurodegenerative diseases.
Metformin, the most widely administered oral anti‑diabetic therapeutic agent, exerts its glucose-lowering effect predominantly via liver kinase B1 (LKB1)-dependent activation of adenosine monophosphate-activated protein kinase (AMPK). Accumulating evidence has demonstrated that metformin possesses potential antitumor effects. However, whether the antitumor effect of metformin is via the LKB1/AMPK signaling pathway remains to be determined. In the current study, the effects of metformin on proliferation, cell cycle progression, and apoptosis of human non‑small cell lung cancer (NSCLC) H460 (LKB1‑null) and H1299 (LKB1‑positive) cells were assessed, and the role of LKB1/AMPK signaling in the anti‑growth effects of metformin were investigated. Cell viability was determined using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, cell cycle distribution and apoptosis were assessed by flow cytometry, and protein expression levels were measured by western blotting. Metformin inhibited proliferation, induced significant cell cycle arrest at the G0‑G1 phase and increased apoptosis in NSCLC cells in a time- and concentration-dependent manner, regardless of the level of LKB1 protein expression. Furthermore, knockdown of LKB1 with short hairpin RNA (shRNA) did not affect the antiproliferative effect of metformin in the H1299 cells. Metformin stimulated AMPK phosphorylation and subsequently suppressed the phosphorylation of mammalian target of rapamycin and its downstream effector, 70‑kDa ribosomal protein S6 kinase in the two cell lines. These effects were abrogated by silencing AMPK with small interfering RNA (siRNA). In addition, knockdown of AMPK with siRNA inhibited the effect of metformin on cell proliferation in the two cell lines. These results provide evidence that the growth inhibition of metformin in NSCLC cells is mediated by LKB1‑independent activation of AMPK, indicating that metformin may be a potential therapeutic agent for the treatment of human NSCLC.
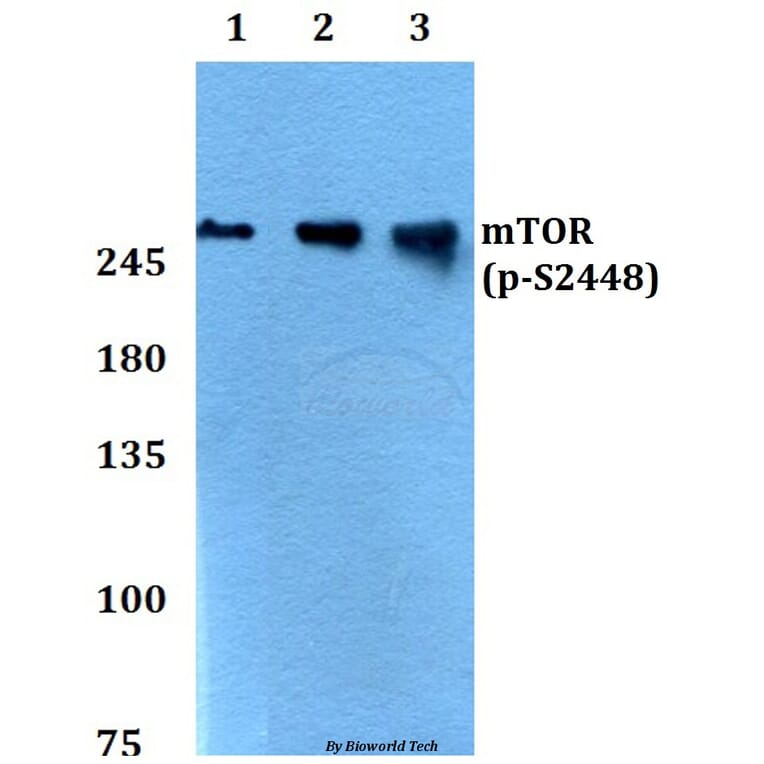
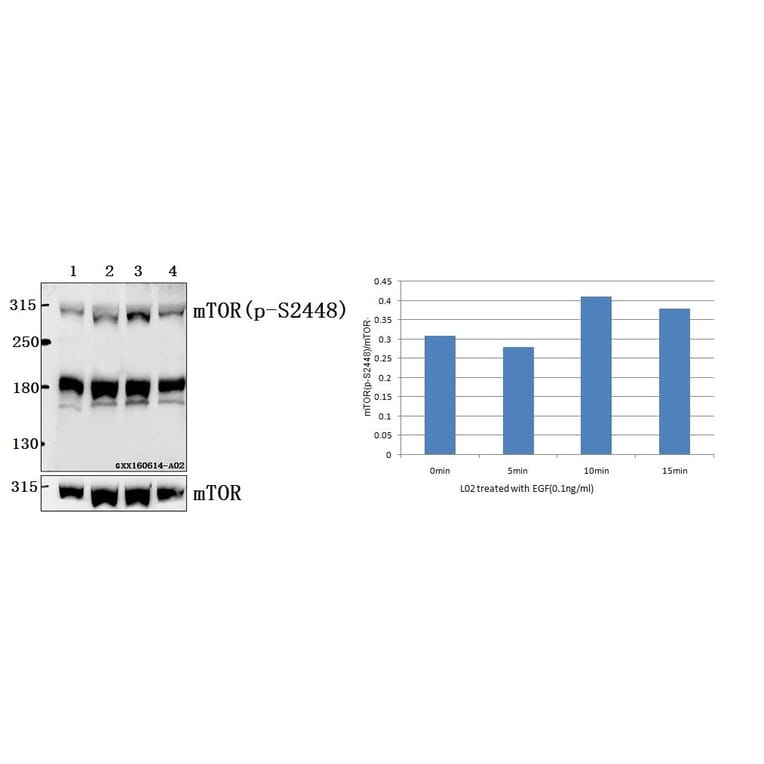


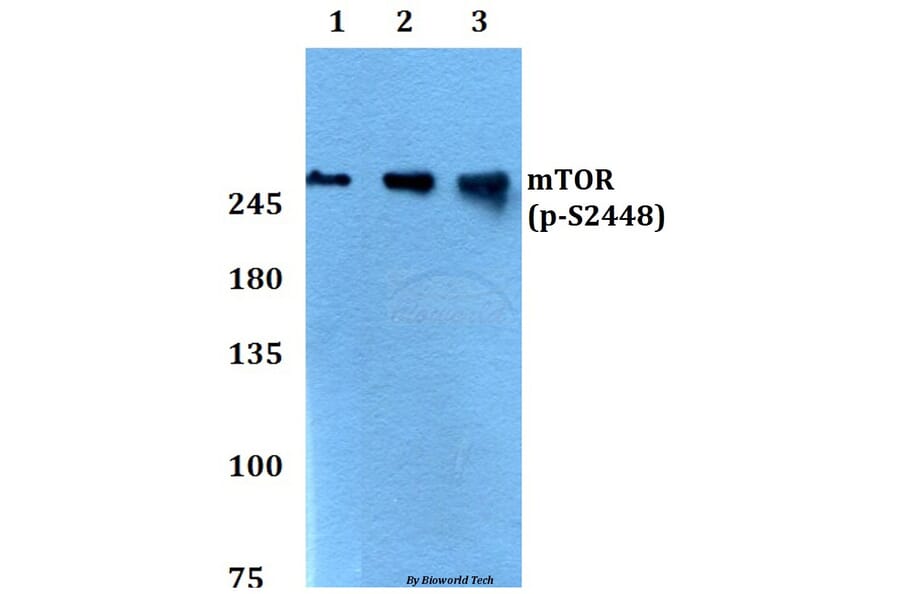
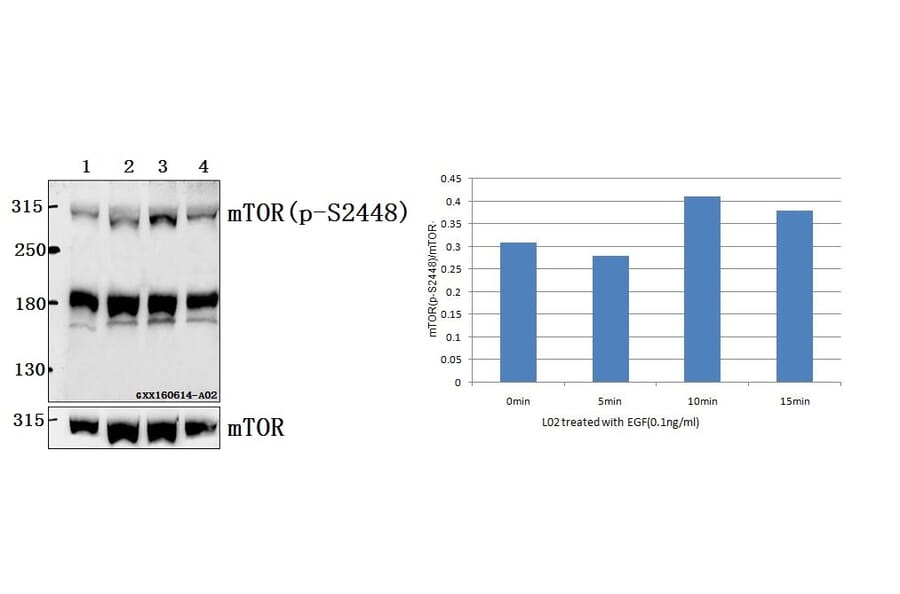

















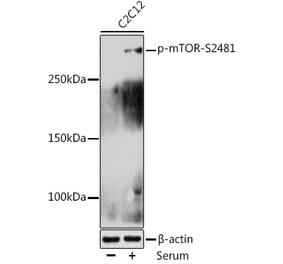
![Western Blot - Anti-mTOR (phospho Ser2448) Antibody [ARC0094] (A308866) - Antibodies.com](https://cdn.antibodies.com/image/catalog/308/A308866_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-mTOR Antibody [RM274] (A121448) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121387_1.png?profile=product_alternative)