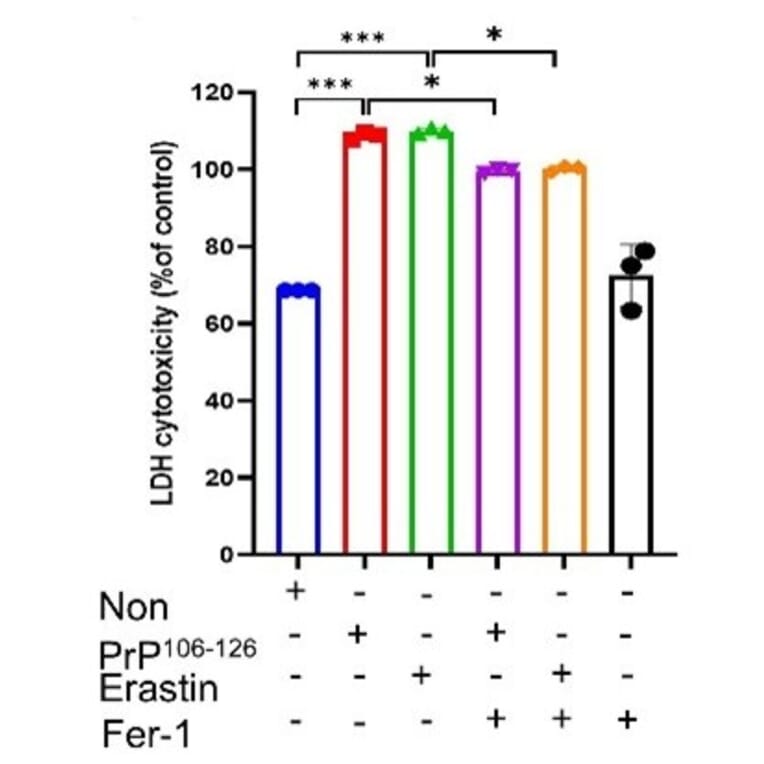
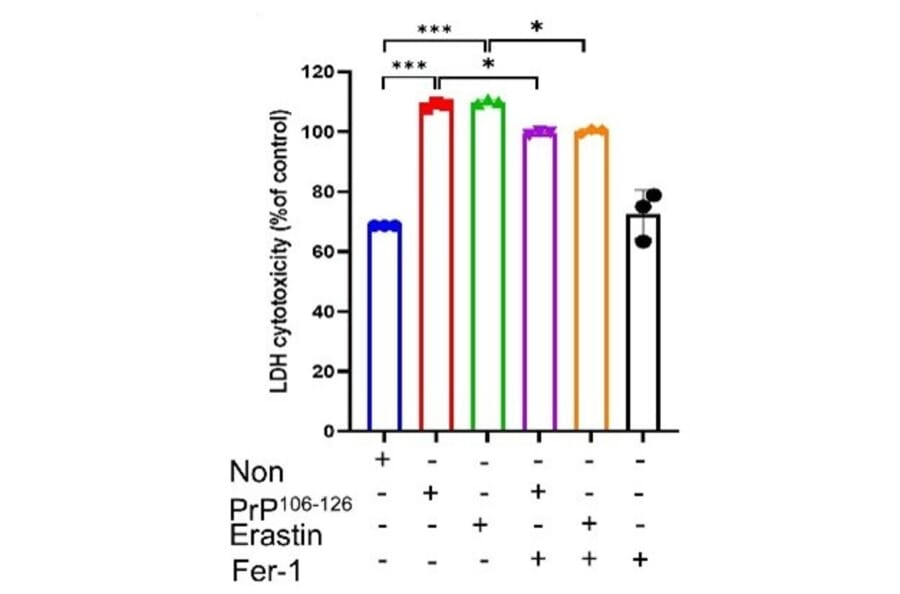
Prion diseases are a group of fatal neurodegenerative disorders caused by misfolded proteins. Understanding the regulatory networks of ferroptosis in prion diseases could unveil new diagnostic and therapeutic strategies. To explore this, we systematically evaluated ferroptosis-associated alterations across human sporadic Creutzfeldt-Jakob disease (sCJD) brain samples, the ME7-infected mouse model, and in vitro using PrP106-126-treated SH-SY5Y cells. In sCJD patients, we observed a significant decrease in GPX4 expression, accompanied by elevated lipid peroxidation, as confirmed by malondialdehyde assays. Furthermore, in vitro experiments using PrP106-126-treated cells confirmed that ferroptosis-related mechanisms actively contribute to cell death, characterized by elevated lipid peroxidation, reactive oxygen species, and increased intracellular Fe2+ levels, as well as diminished glutathione activity. Critically, pharmacological inhibition with ferrostatin-1 effectively mitigated this neurotoxicity, consistent with a ferroptosis-related mechanism. To validate these findings in vivo, we demonstrated that ME7-infected mice exhibited significantly lower levels of GPX4 and SLC7A11, which correlated with increased 4-hydroxynonenal and neuronal damage. Finally, bioinformatic analysis of the GSE124571 dataset identified a distinct transcriptomic signature of 130 differentially expressed ferroptosis-related genes in sCJD patients. These results collectively suggest that ferroptosis-associated alterations are involved in prion-associated neurodegeneration, offering valuable pathophysiological insights into disease progression.
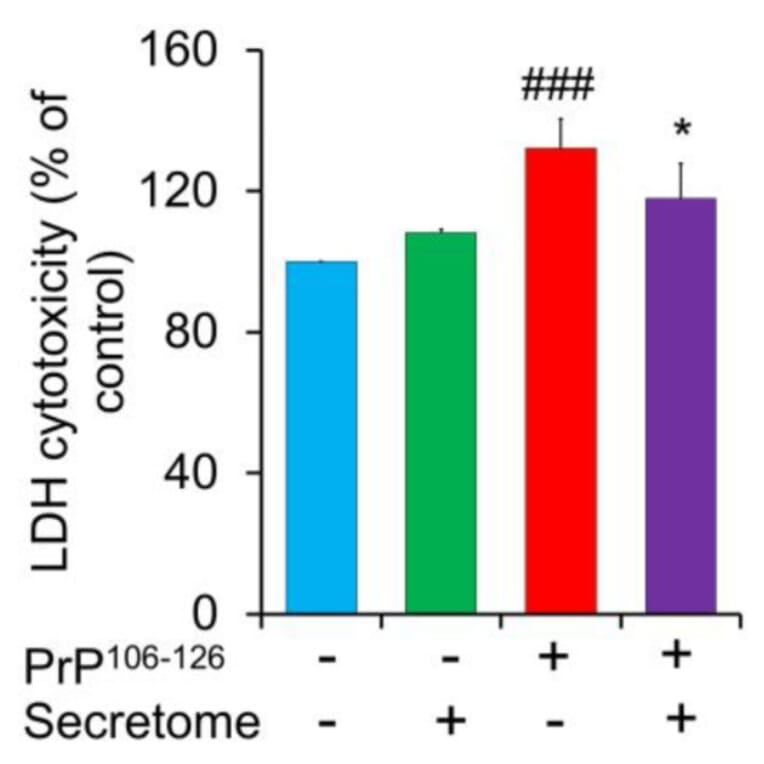
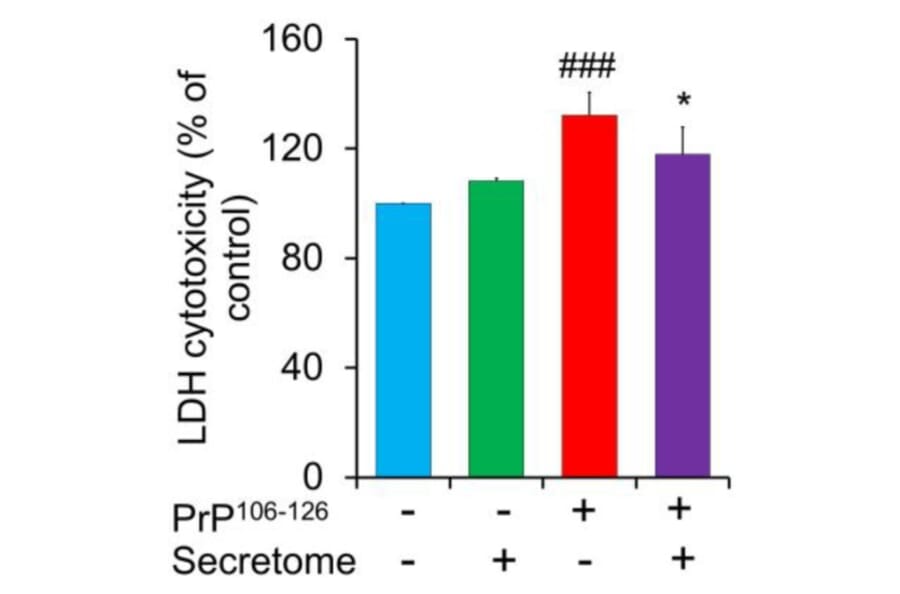
Prion diseases are disorders caused by the misfolding of prion protein (PrPSc), leading to the accumulation of an abnormal form of the normal prion protein (PrP) found in the host. The secretome of mesenchymal stem cells (MSCs), including paracrine-soluble factors, holds promising potential to stimulate host regenerative capability and alleviate organ disorders. In this research, our goal was to investigate the neuroprotective properties of the secretome derived from adipose-derived mesenchymal stem cells (AdMSC secretome) in relation to the toxicity caused by PrP106-126 in SH-SY5Y cells. The findings showed that PrP106-126 treatment exacerbated the neurotoxicity of SH-SY5Y cells, as indicated by increased lactate dehydrogenase (LDH) release. However, the AdMSC secretome significantly decreased LDH release. Under PrP106-126 stimulation, the AdMSC secretome downregulated inflammatory markers (TNF-a and IL-1ß) and upregulated anti-inflammatory IL-10. Treatment with the AdMSC secretome markedly reduced GFAP immunoreactivity in astrocytic C8D1A cells compared to treatment with PrP106-126 alone. In addition, the AdMSC secretome reduced Iba-1 immunoreactivity in BV2 cells activated by LPS. Western blot analysis showed that the AdMSC secretome inhibited pro-apoptotic factor Bax induced by PrP106-126 and increased the expression of anti-apoptotic factor Bcl-2. However, no significant difference was observed in the expression of caspase-3. The AdMSC secretome exhibited a considerable migratory effect on SH-SY5Y cells after 24 h, as demonstrated by the scratch assay. The results suggest that the AdMSC secretome can attenuate PrP106-126-induced neuronal damage.