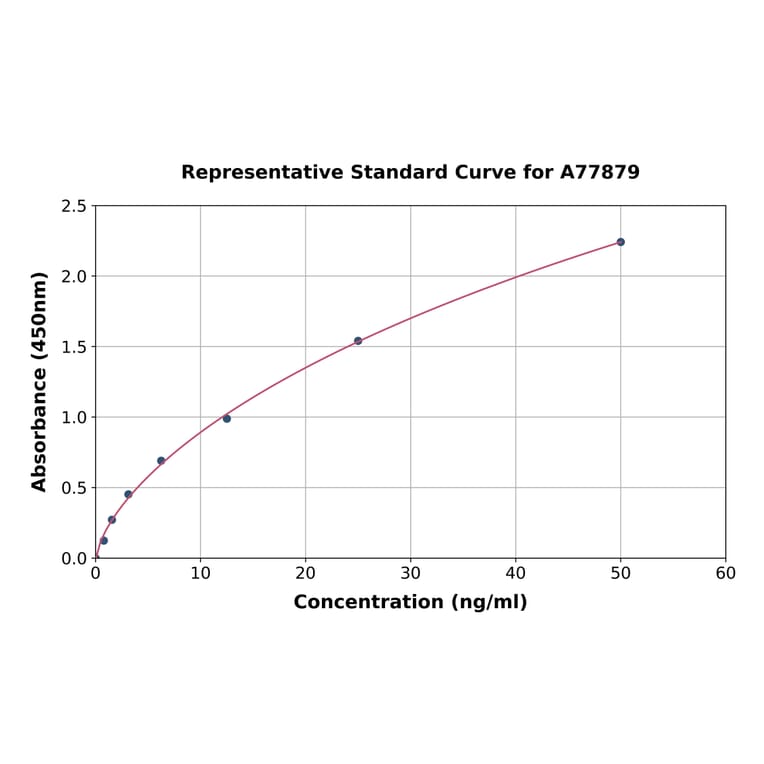
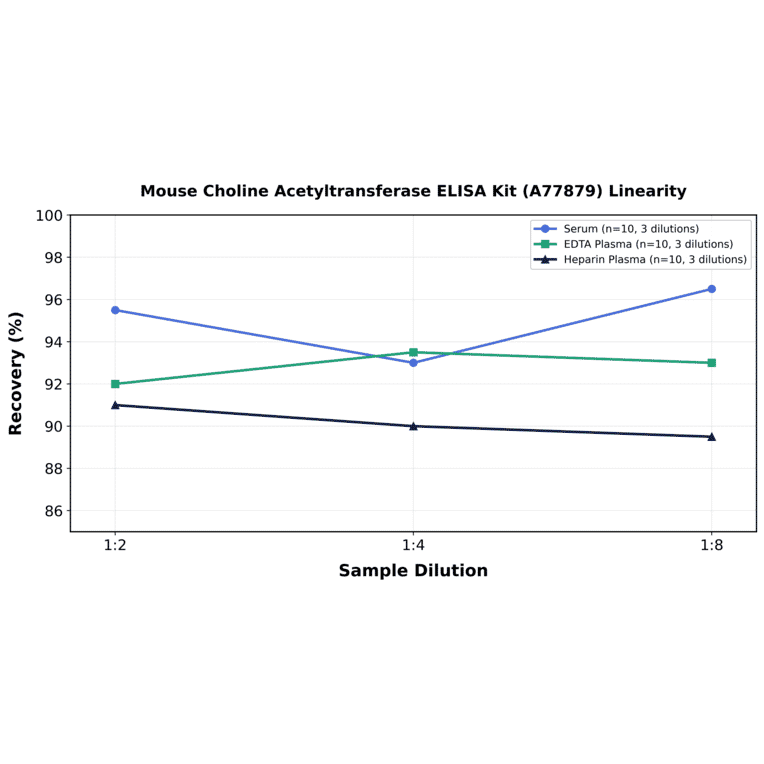
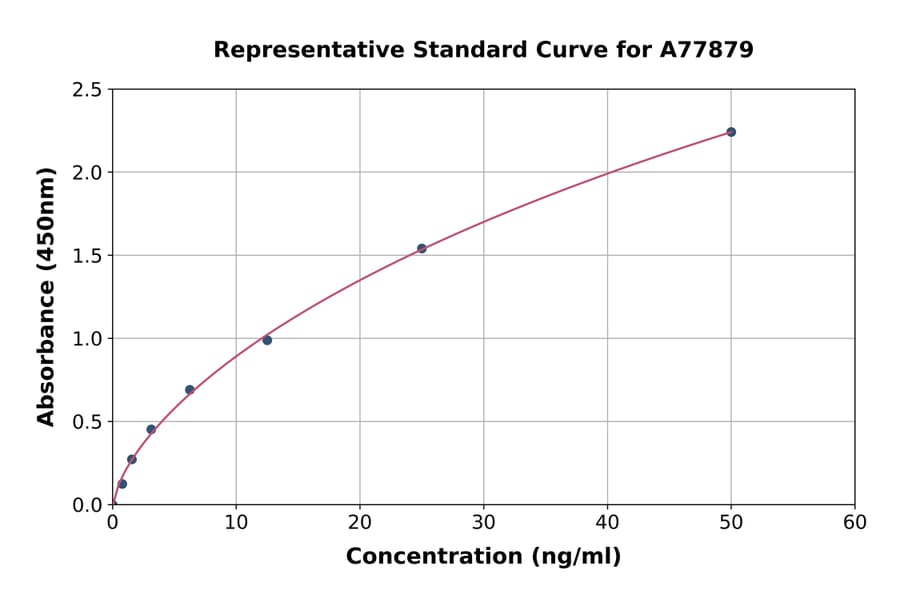
| Sample Type | n | Range | Average |
|---|---|---|---|
| Serum | 10 | 86% - 104% | 98% |
| EDTA Plasma | 10 | 85% - 104% | 94% |
| Heparin Plasma | 10 | 88% - 102% | 96% |
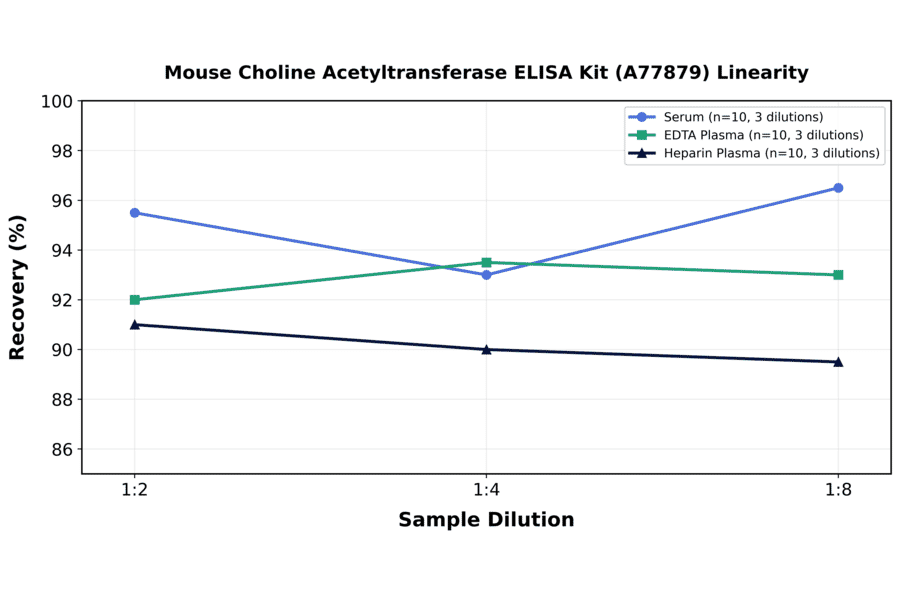
| Sample Type | n | 1:2 | 1:4 | 1:8 |
|---|---|---|---|---|
| Serum | 10 | 90-101% | 86-100% | 91-102% |
| EDTA Plasma | 10 | 84-100% | 86-101% | 85-101% |
| Heparin Plasma | 10 | 82-100% | 83-97% | 87-92% |
| Item | Quantity | Storage |
|---|---|---|
| Pre-Coated 96 Well Microplate | 12 x 8 Well Strips | +4°C |
| Lyopholized Standard | 2 Vials | +4°C |
| Sample Dilution Buffer | 20ml | +4°C |
| Biotinylated Detection Antibody | 120µl | +4°C |
| Antibody Dilution Buffer | 10ml | +4°C |
| HRP-Streptavidin Conjugate | 120µl | +4°C |
| SABC Dilution Buffer | 10ml | +4°C |
| TMB Substrate | 10ml | +4°C |
| Stop Solution | 10ml | +4°C |
| Wash Buffer (25X) | 30ml | +4°C |
| Plate Sealers | 5 Adhesive Strips | - |
| Foil Pouch | 1 Zip-Sealed Pouch | - |
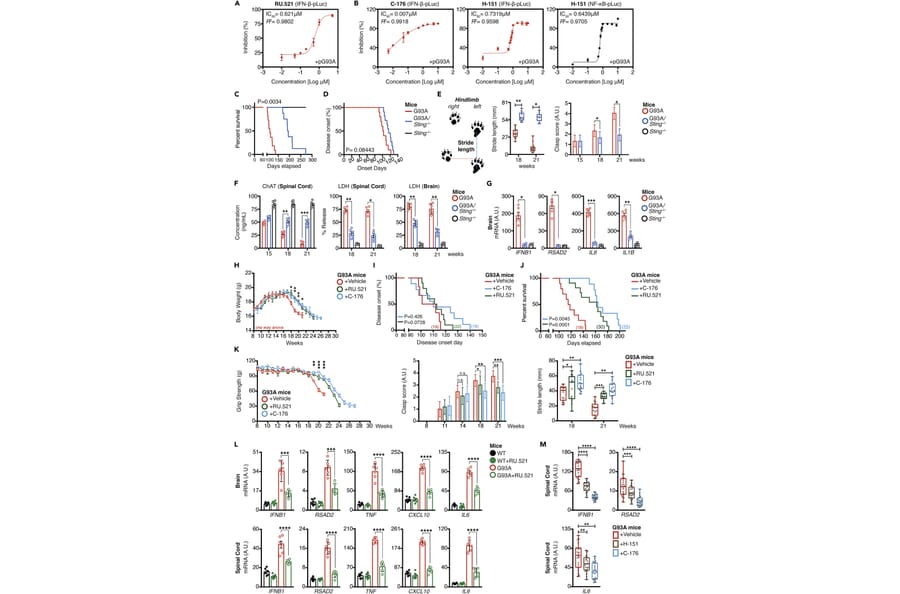
Neuroinflammation exacerbates the progression of SOD1-driven amyotrophic lateral sclerosis (ALS), although the underlying mechanisms remain largely unknown. Herein, we demonstrate that misfolded SOD1 (SOD1Mut)-causing ALS results in mitochondrial damage, thus triggering the release of mtDNA and an RNA:DNA hybrid into the cytosol in an mPTP-independent manner to activate IRF3- and IFNAR-dependent type I interferon (IFN-I) and interferon-stimulating genes. The neuronal hyper-IFN-I and pro-inflammatory responses triggered in ALS-SOD1Mut were sufficiently robust to cause a strong physiological outcome in vitro and in vivo. cGAS/DDX41-STING-signaling is amplified in bystander cells through inter-neuronal gap junctions. Our results highlight the importance of a common DNA-sensing pathway between SOD1 and TDP-43 in influencing the progression of ALS.