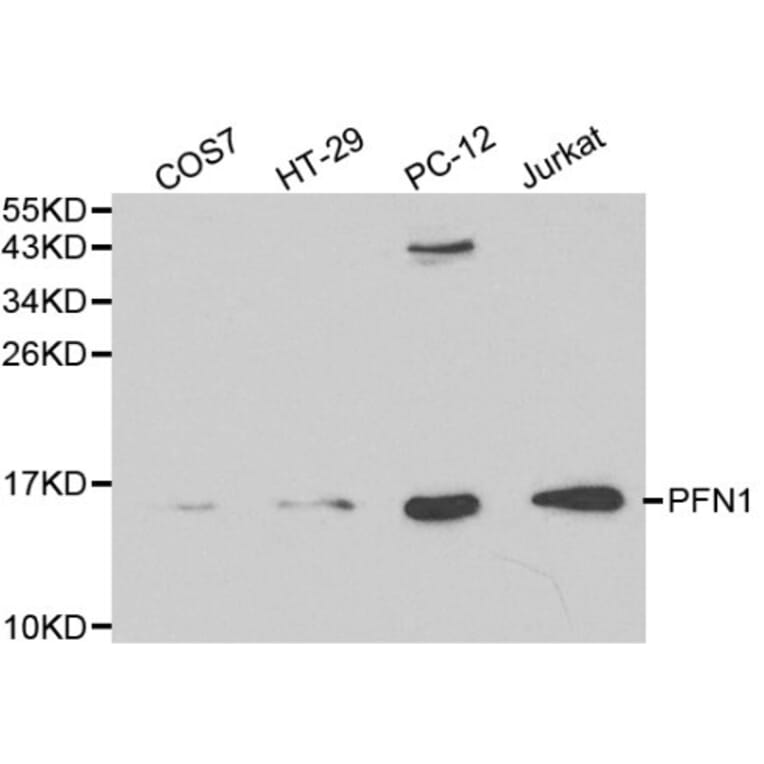
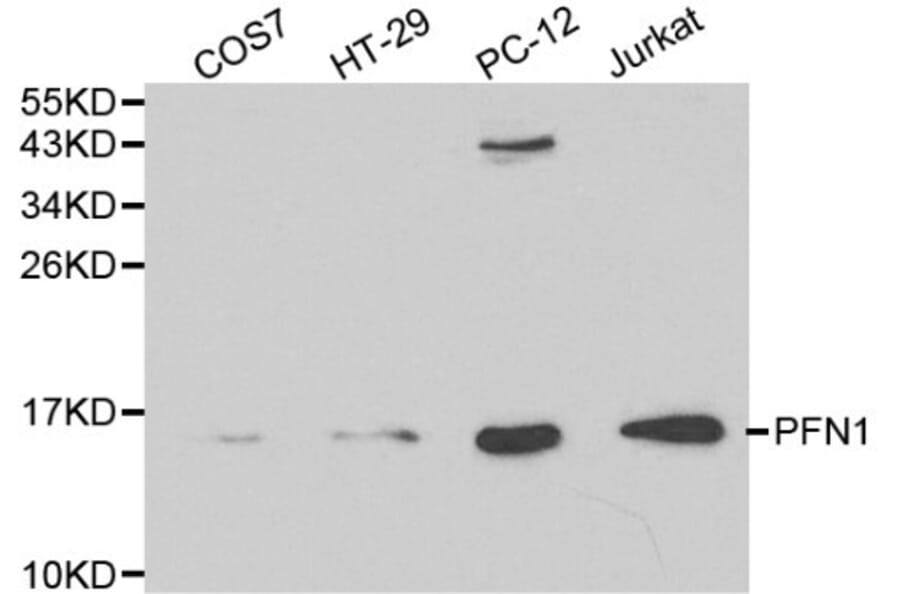
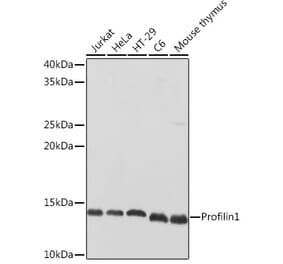
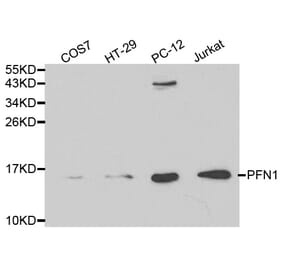
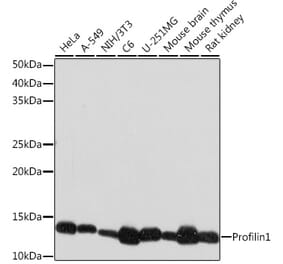
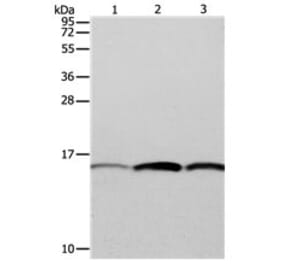
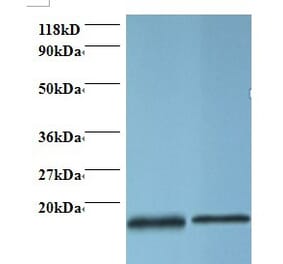
PFN1 pAb detects endogenous levels of PFN1 protein.
Applications
WB, IHC
Reactivity
Human, Mouse, Rat
Immunogen
Recombinant full length Human PFN1.
Host
Rabbit
Clonality
Polyclonal
Conjugate
Unconjugated
Molecular Weight
~ 15 kDa
Purity
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen and the purity is > 95% (by SDS-PAGE).
Product Form
1mg/ml in PBS with 0.1% Sodium Azide, 50% Glycerol.
Synonyms
Epididymis tissue protein Li 184a, Profilin I, Profilin-1