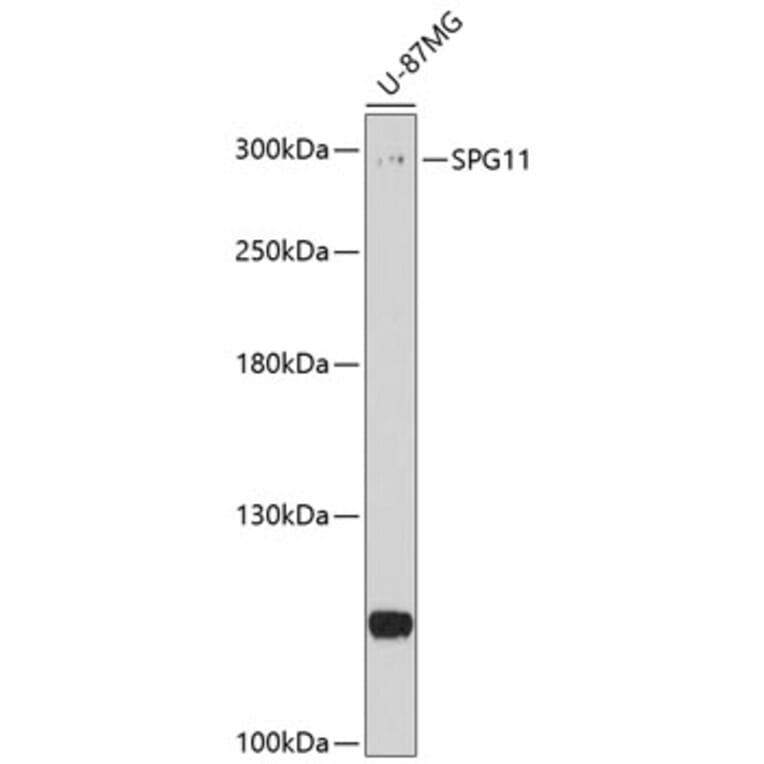
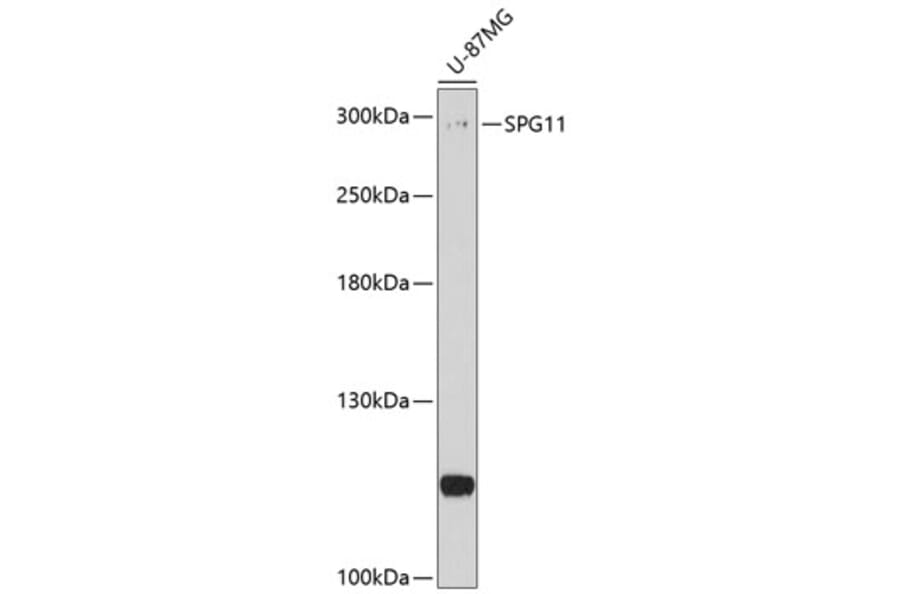
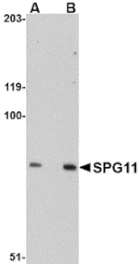
Anti-SPG11 Antibody (A8582) has been discontinued and is no longer available.
View all SPG11 Antibodies.
Unconjugated