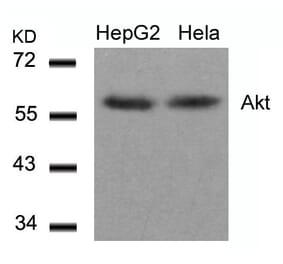
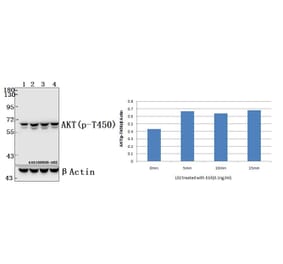
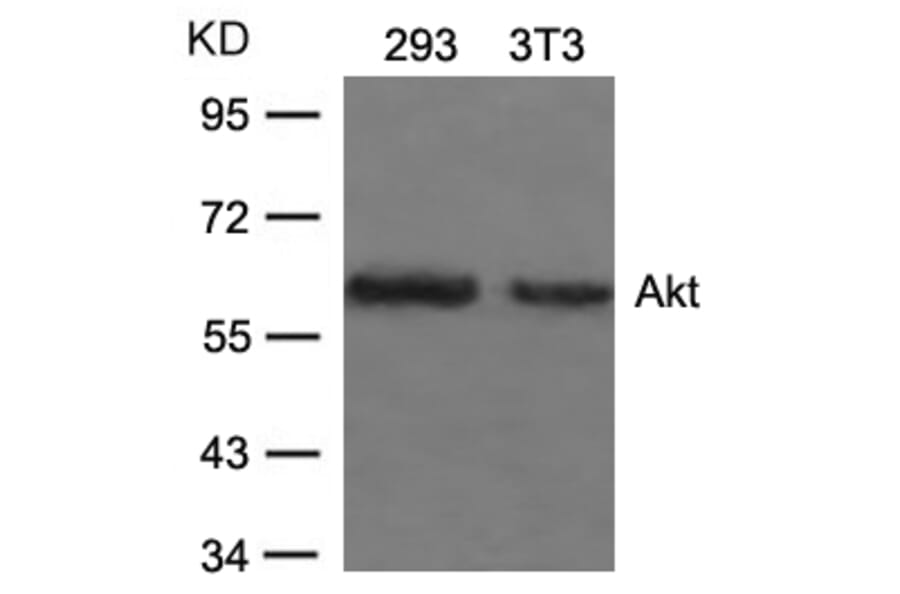
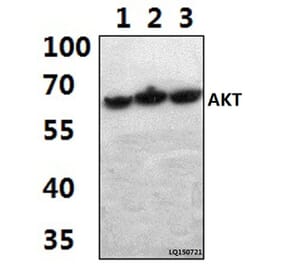
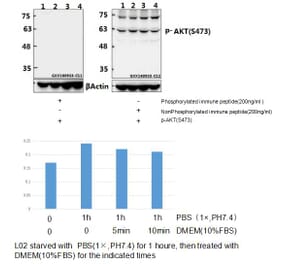
The antibody detects endogenous level of total Akt protein.
Applications
WB, IHC, ICC/IF
Reactivity
Human, Mouse
Immunogen
Peptide sequence around aa. 471~475 (Q-F-S-Y-S) derived from Human Akt.
Host
Rabbit
Clonality
Polyclonal
Conjugate
Unconjugated
Purification
Antibodies were produced by immunizing rabbits with synthetic peptide and KLH conjugates. Antibodies were purified by affinity-chromatography using epitope-specific peptide.
Concentration
1.0 mg/mL
Formulation
Supplied in phosphate-buffered saline (without Mg²? and Ca²?), pH 7.4, containing 150 mM NaCl, 0.02% sodium azide, and 50% glycerol.
Storage
Shipped at 4°C. Upon delivery aliquot and store at -20°C. Avoid freeze/thaw cycles.
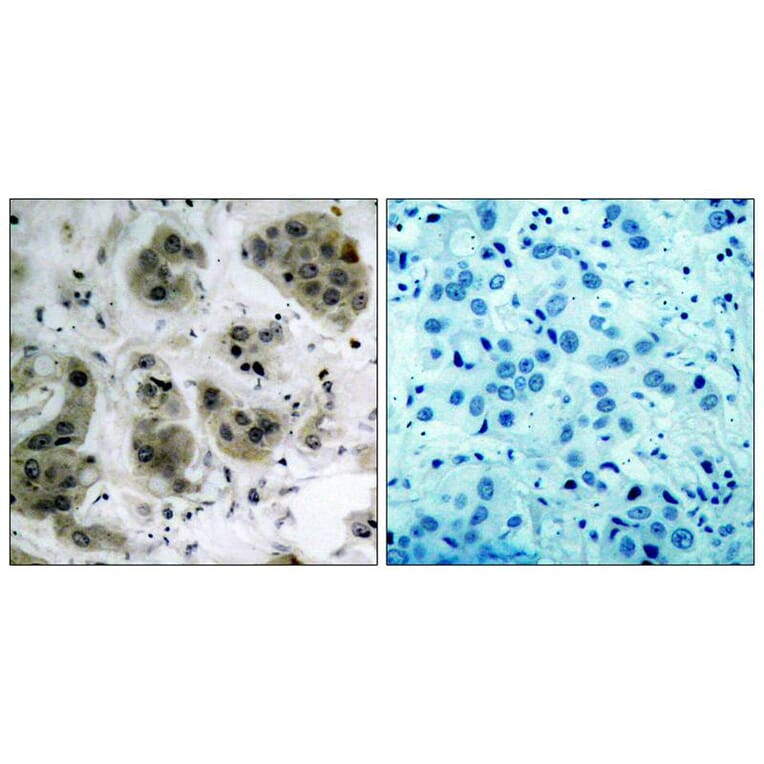
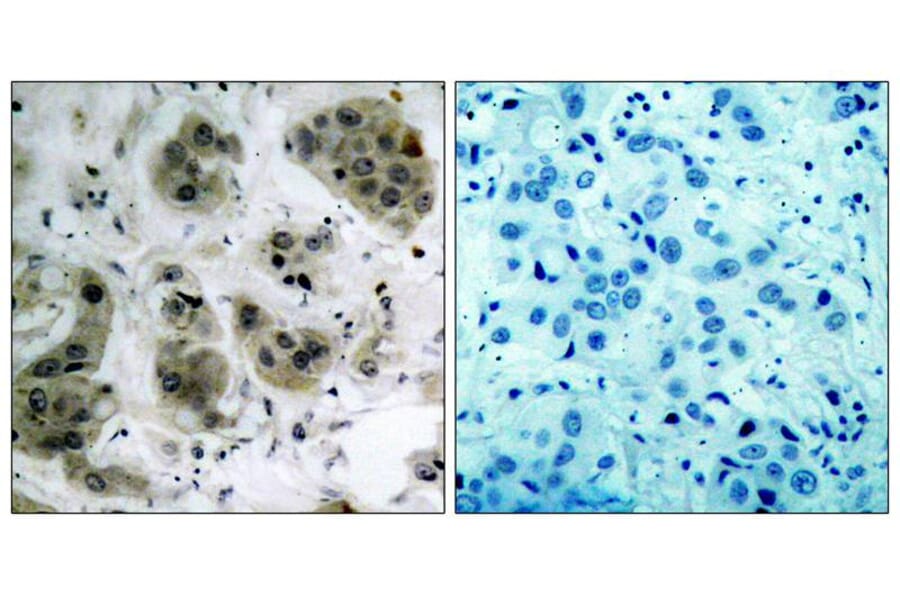
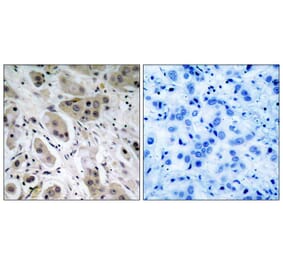
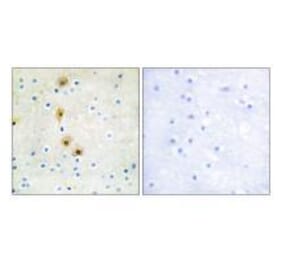
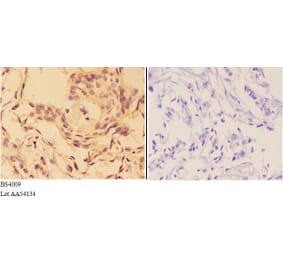
Immunohistochemical analysis of paraffin-embedded human breast carcinoma tissue using Akt (Ab-473) Antibody #21054 (left) or the same antibody preincubated with blocking peptide (right).
Publishing research using Anti-Akt (Ab-473) Antibody (A36519)? Please let us know so that we can list the citation on this page.
Alternative products to Anti-Akt (Ab-473) Antibody (A36519)


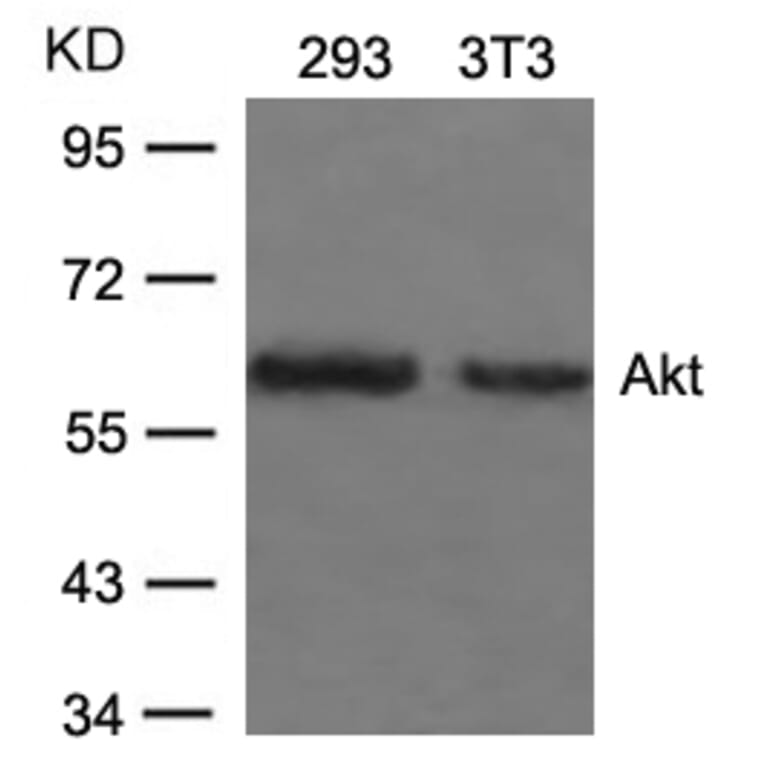


![Immunohistochemistry - Anti-AKT Antibody [RM316] (A121372) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121394_1.png?profile=product_alternative)