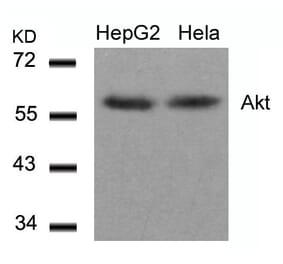
p-AKT (T308) pAb detects endogenous levels of AKT1 only when phosphorylated at Thr308. This antibody also recognizes AKT2 and AKT3 when phosphorylated at the corresponding residues.
Applications
WB, IHC
Reactivity
Human, Mouse, Rat
Immunogen
Synthetic phosphopeptide derived from human AKT1 around the phosphorylation site of Threonine 308.
Host
Rabbit
Clonality
Polyclonal
Conjugate
Unconjugated
Molecular Weight
~ 60 kDa
Purity
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen and the purity is > 95% (by SDS-PAGE).
Product Form
1 mg/ml in Phosphate buffered saline (PBS) with 0.05% sodium azide, approx. pH 7.2.


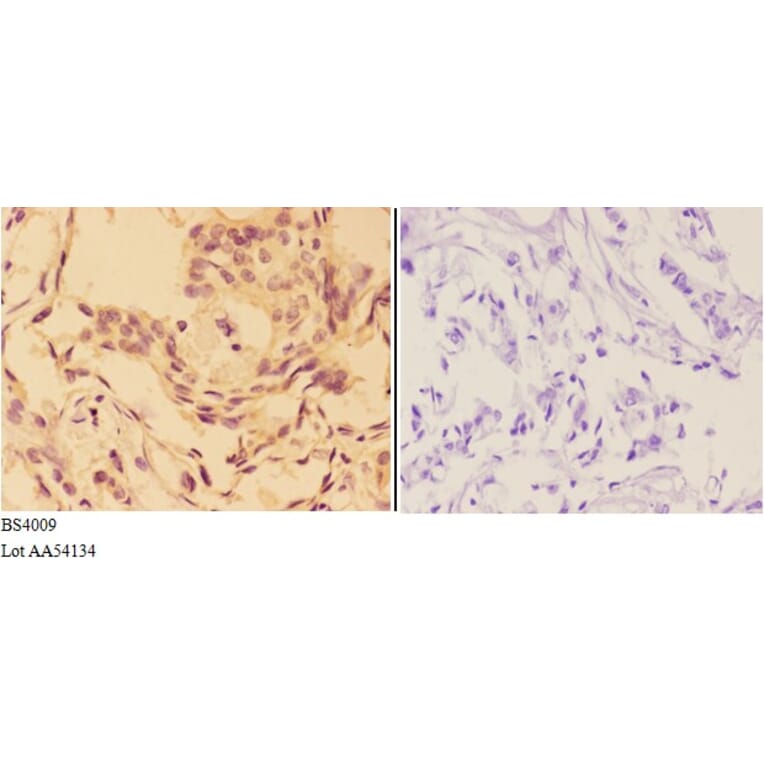
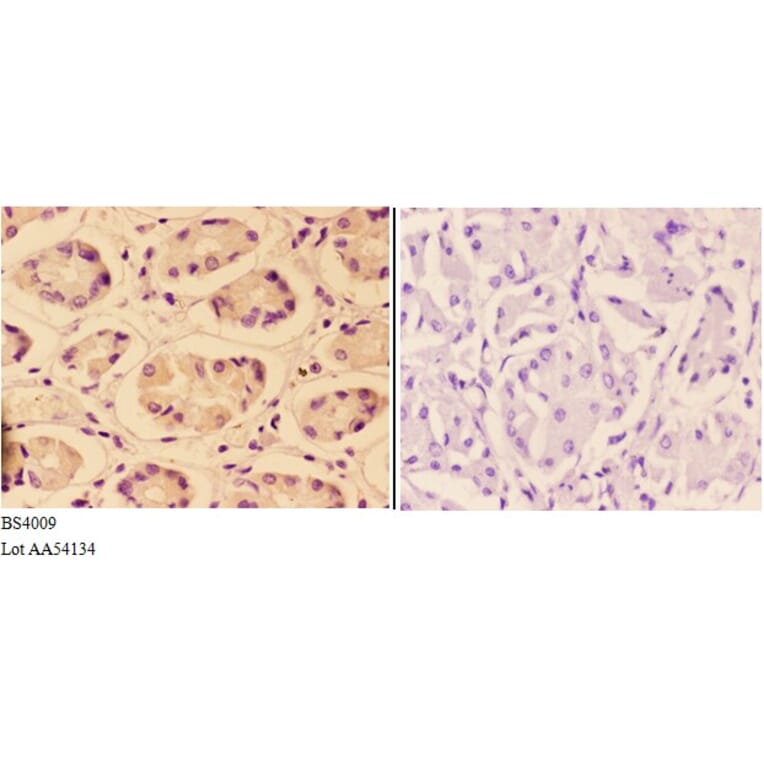


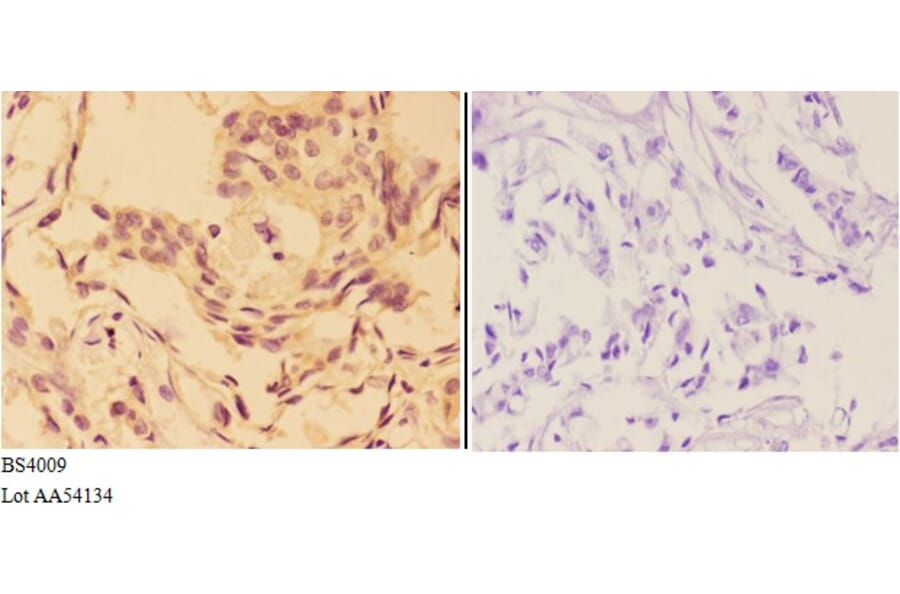
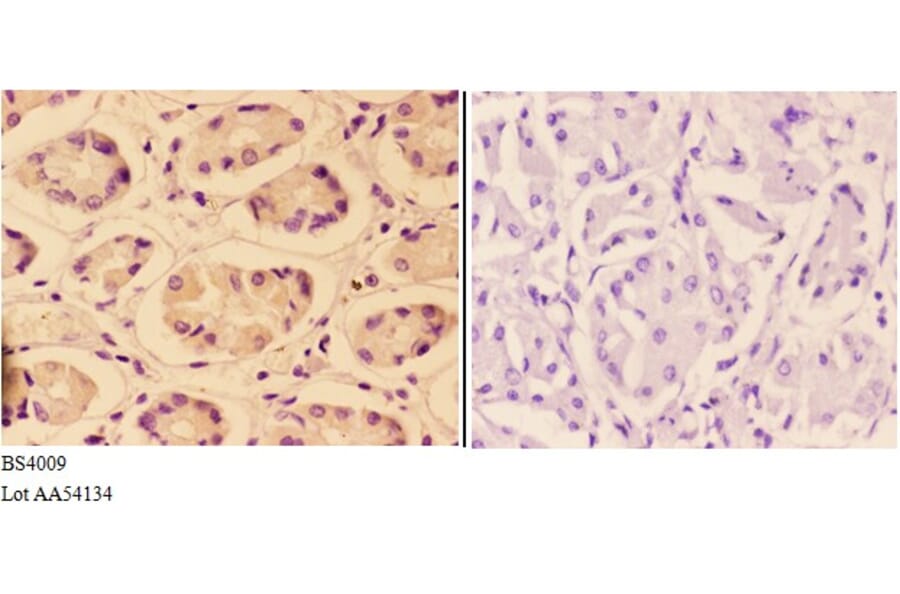
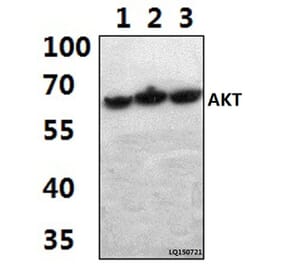
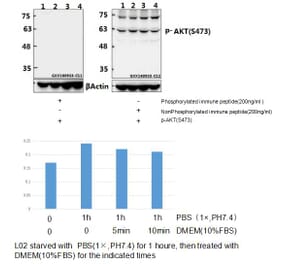
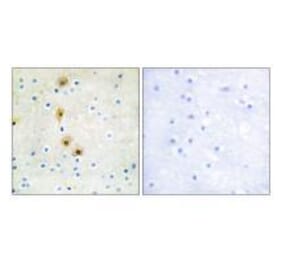
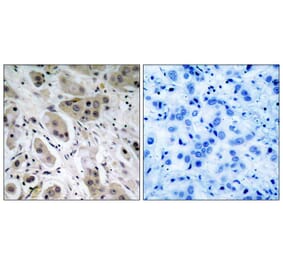



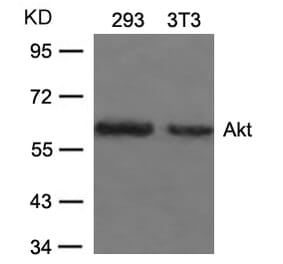

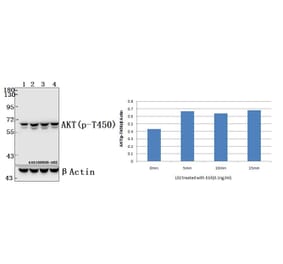
![Immunohistochemistry - Anti-AKT Antibody [RM316] (A121372) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121394_1.png?profile=product_alternative)