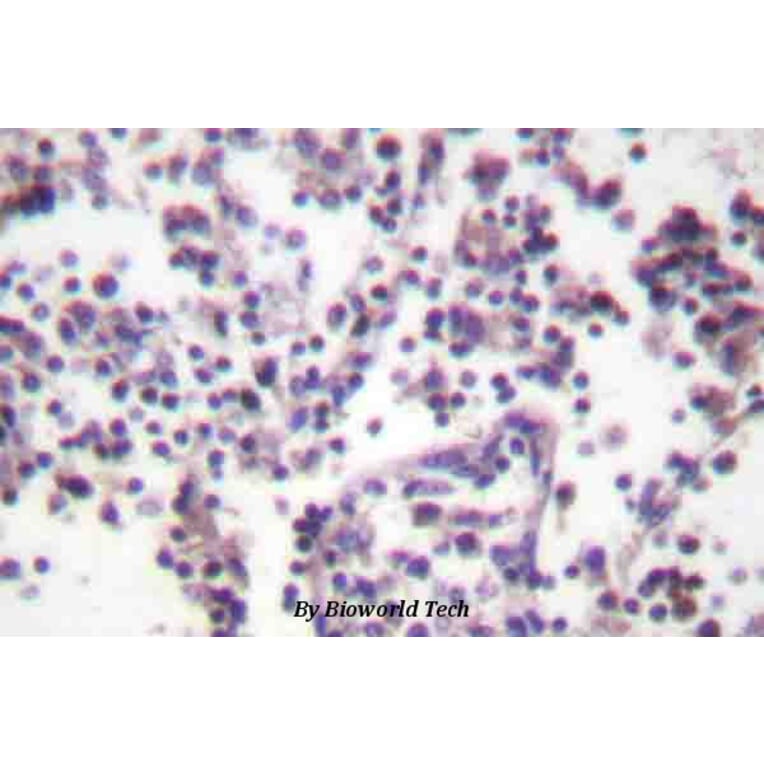
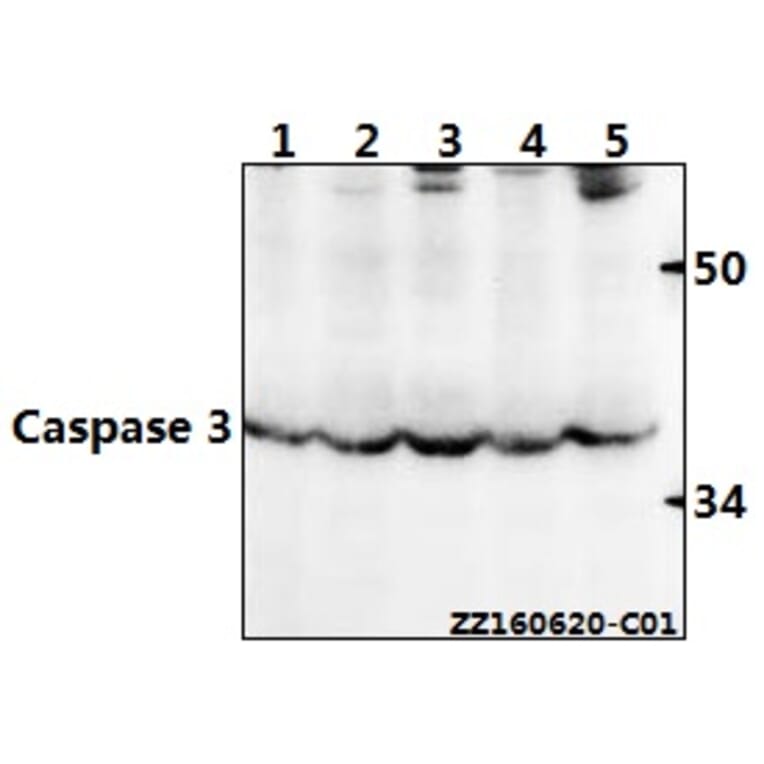
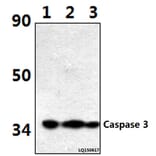
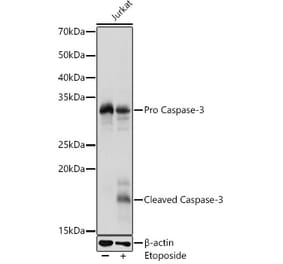
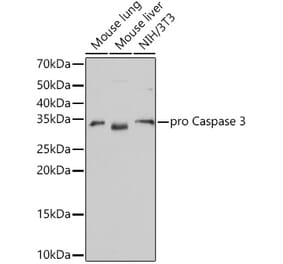
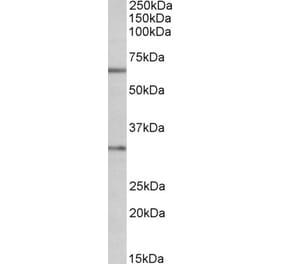
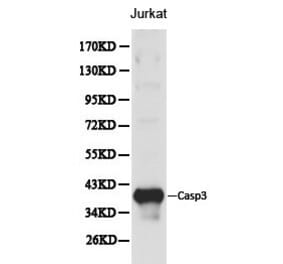
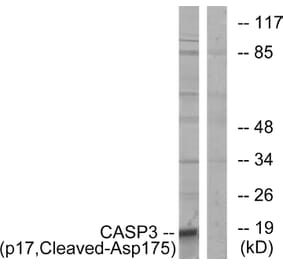
Synthetic peptide, corresponding to amino acids 120-170 of Human Caspase 3.
Host
Rabbit
Clonality
Polyclonal
Conjugate
Unconjugated
Molecular Weight
~ 35 kDa
Purity
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen and the purity is > 95% (by SDS-PAGE).
Product Form
1 mg/ml in Phosphate buffered saline (PBS) with 15 mM sodium azide, approx. pH 7.2.