Primary Antibodies
Secondary Antibodies
Proteins & Peptides
ELISA Kits
About Us
Contact Us
Sign In/Register
0
ISO 9001:2015 Certified
Live Customer Support
4.5/5 on Trustpilot
100% Quality Guarantee
Home
Primary Antibodies
Huntingtin Antibodies
Anti-Huntingtin Antibody (A121602)
Anti-Huntingtin Antibody (A121602)
Overview
Specifications
Images
Enlarge Image
Enlarge Image
$590
Product Datasheet
Goat polyclonal antibody to Huntingtin for WB, IF and IHC-P.
100% Guarantee
Price Match Guarantee
Product Size:
600µg
Quantity:
1
2
3
4
5
6
7
8
9
10
Add To Cart
Request a Quotation
Custom or Bulk Request
Shipping Information
Freight/Packing Charges:
$40
Dispatched from St. Louis, MO.
Lead Time: 5-8 business days.
Specifications
Name
Anti-Huntingtin Antibody
Description
Goat polyclonal antibody to Huntingtin.
Specificity
This antibody recognizes wild type and mutant Huntingtin.
Applications
WB
,
IF
,
IHC-P
Dilutions
WB: 1:500-1:2,000, IF: 1:500-1:2,000, IHC-P: 1:500-1:2,000, IHC-Fr: 1:500-1:2,000
Reactivity
Human, Rat, Mouse
Immunogen
Recombinant peptide derived from within amino acids 85-200 of human Huntingtin, expressed in and purified from E. coli
Host
Goat
Clonality
Polyclonal
Isotype
IgG
Conjugate
Unconjugated
Purification
Affinity purification.
Concentration
3 mg/ml
Product Form
Liquid
Formulation
Supplied in Phosphate Buffered Saline with 20% Glycerol and 0.05% Sodium Azide.
Storage
Shipped at 4°C. Upon delivery aliquot and store at -20°C. Avoid freeze/thaw cycles.
Synonyms
HD, HD protein, HTT, Huntington disease protein, IT15
Isotype Controls
Goat IgG (A121671)
Suitable Secondaries
Donkey Anti-Goat IgG H&L Antibody (AP) (A300679)
Donkey Anti-Goat IgG H&L Antibody (Biotin) (A300716)
Donkey Anti-Goat IgG H&L Antibody (FITC) (A300685)
Donkey Anti-Goat IgG H&L Antibody (HRP) (A300730)
See all Anti-Goat IgG Secondaries →
Disclaimer
This product is for research use only. It is not intended for diagnostic or therapeutic use.
Scientific Validation Data
Validation Data
(2)
Enlarge Image
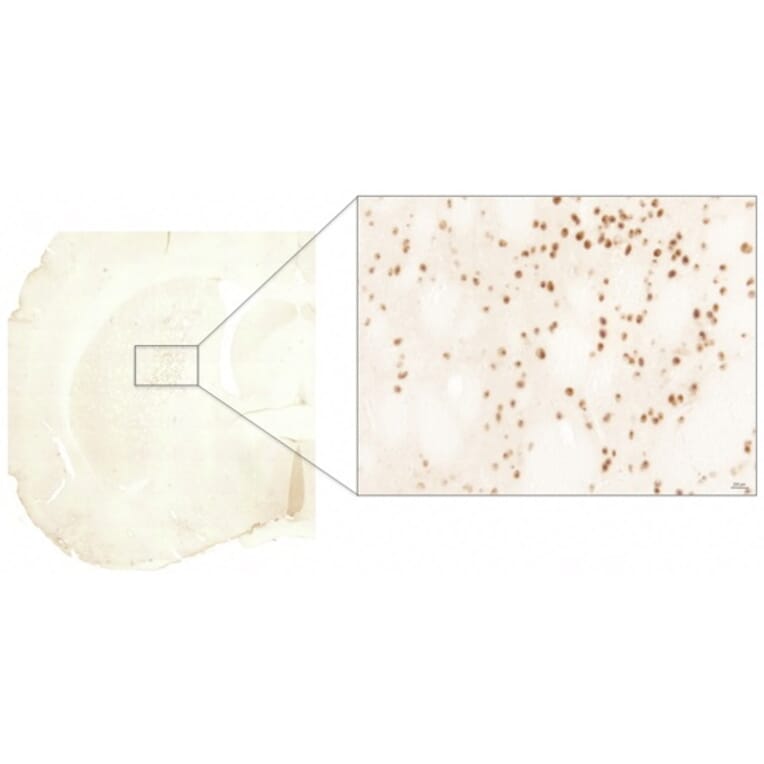
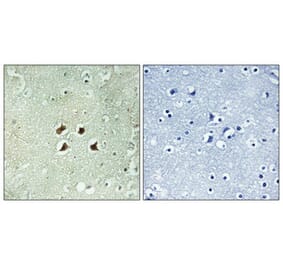
Immunohistochemistry - Anti-Huntingtin Antibody (A121602)
Anti-RAB27A Antibody (1:2,000) staining of rat LV-Htt171-82Q (800 ng), PFA 4%, PBS-BSA 1% – 25µm.
Enlarge Image
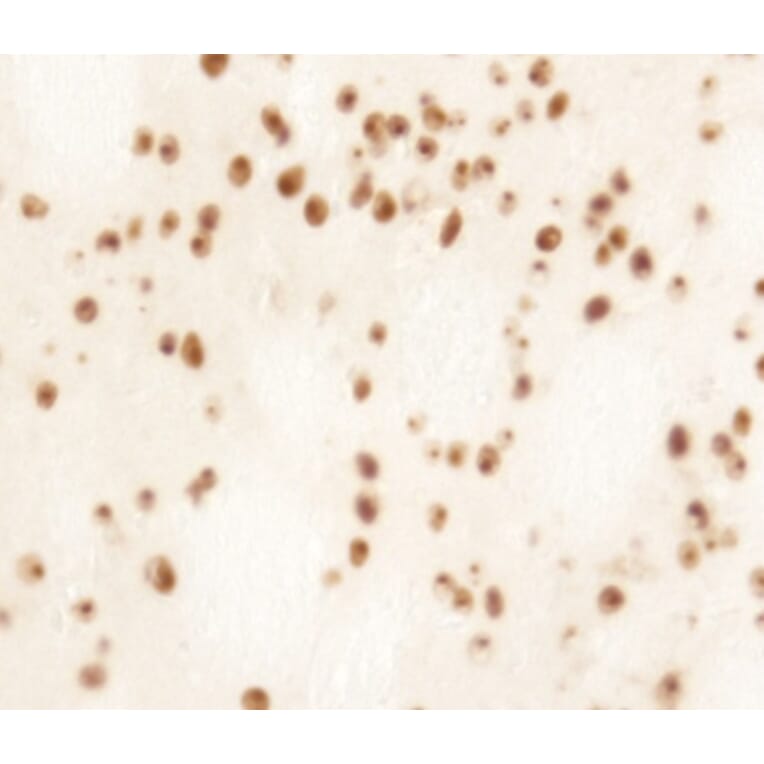
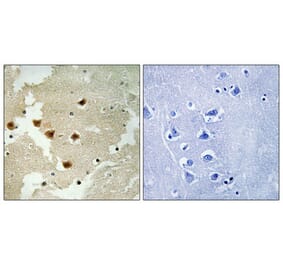
Immunohistochemistry - Anti-Huntingtin Antibody (A121602)
Anti-RAB27A Antibody (1:2,000) staining of rat LV-Htt171-82Q (800 ng), PFA 4%, PBS-BSA 1% – 25µm.
Publishing research using Anti-Huntingtin Antibody (A121602)? Please
let us know
so that we can list the citation on this page.
Alternative products to Anti-Huntingtin Antibody (A121602)
(3)
A308268
Anti-Huntingtin Antibody
Rabbit polyclonal antibody to Huntingtin for WB, IHC and ICC/IF.
A280584
Anti-Huntingtin Antibody [HDB4E10]
Mouse monoclonal [HDB4E10] antibody to Huntingtin for WB, IHC-Fr and IP.
A95558
Anti-Huntingtin Antibody
Rabbit polyclonal antibody to Huntingtin for IHC, IF and ELISA.
A280585
Anti-Huntingtin Antibody [HDC8A4]
Mouse monoclonal [HDC8A4] antibody to Huntingtin for WB, IHC-Fr and IP.
A346673
Anti-Huntingtin Antibody [23GB6285]
Recombinant rabbit monoclonal [23GB6285] antibody to Huntingtin for WB, Flow Cytometry, ICC/IF and IHC-P.
A346672
Anti-Huntingtin Antibody [24GB10980]
Recombinant rabbit monoclonal [24GB10980] antibody to Huntingtin for WB, Flow Cytometry, ICC/IF and IHC-P.
A93948
Anti-Huntingtin (phospho Ser421) Antibody
Rabbit polyclonal antibody to Huntingtin (phospho Ser421) for IHC, IF and ELISA.
A337903
Anti-Huntingtin Nanobody [SAA1178]
Recombinant alpaca monoclonal [SAA1178] nanobody to Huntingtin for DB, ELISA and SPR.
(2)
A337905
Anti-Huntingtin Nanobody [SAA1033]
Recombinant alpaca monoclonal [SAA1033] nanobody to Huntingtin for ELISA.
A337904
Anti-Huntingtin Nanobody [SAA1032]
Recombinant alpaca monoclonal [SAA1032] nanobody to Huntingtin for ELISA.
(3)
A308268
Anti-Huntingtin Antibody
Rabbit polyclonal antibody to Huntingtin for WB, IHC and ICC/IF.
A280584
Anti-Huntingtin Antibody [HDB4E10]
Mouse monoclonal [HDB4E10] antibody to Huntingtin for WB, IHC-Fr and IP.
A95558
Anti-Huntingtin Antibody
Rabbit polyclonal antibody to Huntingtin for IHC, IF and ELISA.
A280585
Anti-Huntingtin Antibody [HDC8A4]
Mouse monoclonal [HDC8A4] antibody to Huntingtin for WB, IHC-Fr and IP.
A346673
Anti-Huntingtin Antibody [23GB6285]
Recombinant rabbit monoclonal [23GB6285] antibody to Huntingtin for WB, Flow Cytometry, ICC/IF and IHC-P.
A346672
Anti-Huntingtin Antibody [24GB10980]
Recombinant rabbit monoclonal [24GB10980] antibody to Huntingtin for WB, Flow Cytometry, ICC/IF and IHC-P.
A93948
Anti-Huntingtin (phospho Ser421) Antibody
Rabbit polyclonal antibody to Huntingtin (phospho Ser421) for IHC, IF and ELISA.
A337903
Anti-Huntingtin Nanobody [SAA1178]
Recombinant alpaca monoclonal [SAA1178] nanobody to Huntingtin for DB, ELISA and SPR.
(2)
A337905
Anti-Huntingtin Nanobody [SAA1033]
Recombinant alpaca monoclonal [SAA1033] nanobody to Huntingtin for ELISA.
A337904
Anti-Huntingtin Nanobody [SAA1032]
Recombinant alpaca monoclonal [SAA1032] nanobody to Huntingtin for ELISA.
See all Huntingtin Antibodies
Proteins predicted to interact with Huntingtin
Predicted protein interactions based upon String database. Revelancy score correlates with probability of interaction.
Huntingtin Associated Protein 1 Antibodies
99.9% Relevancy Score
p53 Antibodies
99.8% Relevancy Score
CBP Antibodies
99.7% Relevancy Score
Optineurin Antibodies
99.7% Relevancy Score
IP3 Receptor Antibodies
99.5% Relevancy Score
ZDHHC17 Antibodies
99.5% Relevancy Score
GAPDH Antibodies
99.3% Relevancy Score
REST Antibodies
99.1% Relevancy Score
FNBP3 Antibodies
98.9% Relevancy Score
Top





![Western Blot - Anti-Huntingtin Antibody [HDB4E10] (A280584) - Antibodies.com](https://cdn.antibodies.com/image/catalog/280/A280584_1.jpg?profile=product_alternative)

![SDS-PAGE - Anti-Huntingtin Nanobody [SAA1178] (A337903) - Antibodies.com](https://cdn.antibodies.com/image/catalog/337/A337903_1.jpg?profile=product_alternative)
![SDS-PAGE - Anti-Huntingtin Nanobody [SAA1033] (A337905) - Antibodies.com](https://cdn.antibodies.com/image/catalog/337/A337905_1.jpg?profile=product_alternative)
![SDS-PAGE - Anti-Huntingtin Nanobody [SAA1032] (A337904) - Antibodies.com](https://cdn.antibodies.com/image/catalog/337/A337904_1.jpg?profile=product_alternative)