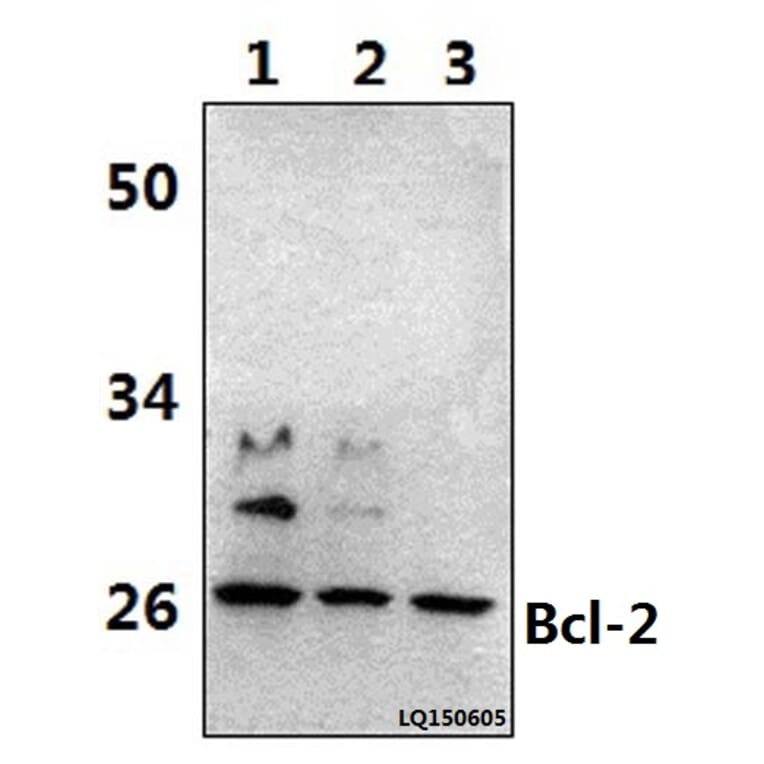
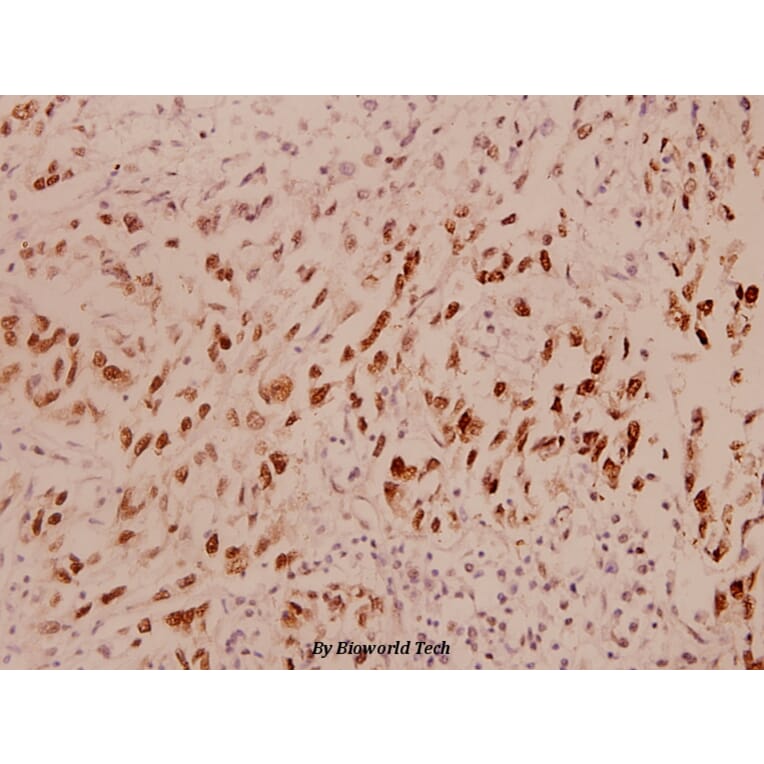
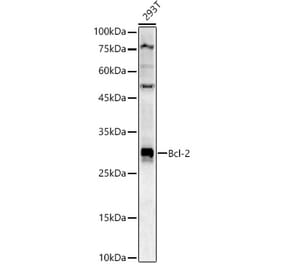
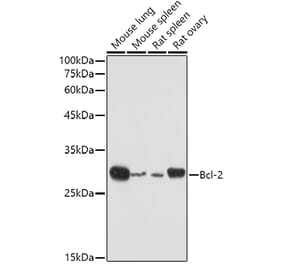
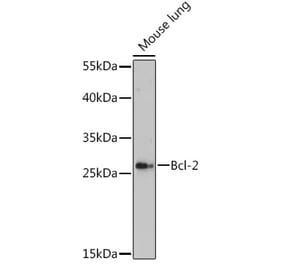
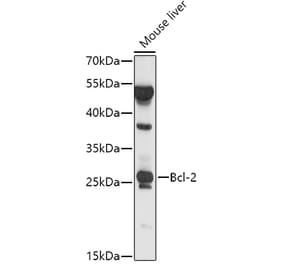
Anti-Bcl-2 (D64) Antibody (A25061) has been discontinued and is no longer available.
View all Anti-Bcl-2 Antibodies.

Unconjugated
A series of 12 novel acylhydrazone, chalcone and amide-bridged analogues of combretastatin A-4 were designed and synthesized toward tubulin. All these compounds were determined by elemental analysis, (1)H NMR, and MS. Among them, compound 7 with acylhydrazone-bridge, bearing a benzyl at the indole-N position, was identified as a potent antiproliferative agent against a panel of cancer cell lines with IC50 values ranging from 0.08 to 35.6 μM. In contrast, its cytotoxic effects on three normal human cells were minimal. Cellular studies have revealed that the induction of apoptosis by compound 7 was associated with a collapse of mitochondrial membrane potential, accumulation of reactive oxygen species, alterations in the expression of some cell cycle-related proteins (Cyclin B1, Cdc25c, Cdc2, P21) and some apoptosis-related proteins (Bax, PARP, Bcl-2, Caspase3). The docking mode showed the binding posture of CA-4 and compound 7 are similar in the colchicine-binding pocket of tubulin, as confirmed by colchicine-tubulin competitive binding assay, tubulin polymerization inhibitory activity, extracellular protein expression determination assay and confocal immunofluorescence microscopy. In vivo study, compound 7 effectively inhibited A549 xenograft tumor growth without causing significant loss of body weight suggesting that compound 7 is a promising new antimitotic agent with clinical potential.
Emerging evidence indicates that methylglyoxal (MG) can inhibit tumorigenesis. Glyoxalase I (GLOI), a MG degradation enzyme, is implicated in the progression of human malignancies. However, little is known about the roles of MG and GLOI in breast cancer. Our purpose was to investigate the anticancer effects of MG and inhibition of GLOI on breast cancer cells and the underlying mechanisms of these effects. Our findings demonstrate that cell viability, migration, invasion, colony formation, and tubule formation were significantly restrained by addition of MG or inhibition of GLOI, while apoptosis was significantly increased. Furthermore, the expression of p-JNK, p-ERK, and p-p38 was markedly upregulated by addition of MG or inhibition of GLOI, whereas MMP-9 and Bcl-2 expression levels were dramatically decreased. These effects were augmented by combined treatment with MG and inhibition of GLOI. Collectively, these data indicate that MG or inhibition of GLOI induces anticancer effects in breast cancer cells and that these effects are potentiated by combination of the 2. These effects were modulated by activation of the MAPK family and downregulation of Bcl-2 and MMP-9. These findings may provide a new approach for the treatment of breast cancer.
Interleukin-33 (IL-33) plays a protective role in myocardial ischemia and reperfusion (I/R) injury, but the underlying mechanism was not fully elucidated. The present study was designed to investigate whether IL-33 protects against myocardial I/R injury by regulating both P38 mitogen-activated-protein kinase (P38 MAPK), which is involved in one of the downstream signaling pathways of IL-33, and high mobility group box protein 1 (HMGB1), a late pro-inflammatory cytokine. Myocardial I/R injury increased the level of IL-33 and its induced receptor (sST) in myocardial tissue. Compared with the I/R group, the IL-33 group had significantly lower cardiac injury (lower serum creatine kinase (CK), lactate dehydrogenase (LDH), and cTnI levels and myocardial infarct size), a suppressed inflammatory response in myocardial tissue (lower expression of HMGB1, IL-6, TNF-α and INF-γ) and less myocardial apoptosis (much higher Bcl-2/Bax ratio and lower cleaved caspase-3 expression). Moreover, IL-33 activated the P38 MAPK signaling pathway (up-regulating P-P38 expression) in myocardial tissue, and SB230580 partially attenuated the anti-inflammatory and anti-apoptosis effects of IL-33. These findings indicated that IL-33 protects against myocardial I/R injury by inhibiting inflammatory responses and myocardial apoptosis, which may be associated with the HMGB1 and P38 MAPK signaling pathways.
Probiotics actively participate in neuropsychiatric disorders. However, the role of gut microbiota in brain disorders and vascular dementia (VaD) remains unclear. We used a mouse model of VaD induced by a permanent right unilateral common carotid arteries occlusion (rUCCAO) to investigate the neuroprotective effects and possible underlying mechanisms of Clostridium butyricum. Following rUCCAO, C. butyricum was intragastrically administered for 6 successive weeks. Cognitive function was estimated. Morphological examination was performed by electron microscopy and hematoxylin-eosin (H&E) staining. The BDNF-PI3K/Akt pathway-related proteins were assessed by western blot and immunohistochemistry. The diversity of gut microbiota and the levels of butyrate in the feces and the brains were determined. The results showed that C. butyricum significantly attenuated the cognitive dysfunction and histopathological changes in VaD mice. C. butyricum not only increased the levels of BDNF and Bcl-2 and decreased level of Bax but also induced Akt phosphorylation (p-Akt) and ultimately reduced neuronal apoptosis. Moreover, C. butyricum could regulate the gut microbiota and restore the butyrate content in the feces and the brains. These results suggest that C. butyricum might be effective in the treatment of VaD by regulating the gut-brain axis and that it can be considered a new therapeutic strategy against VaD.
Phloretin (Ph) existing in apples, pears and various vegetables is known to have antitumor activities in several cancer cell lines. However, little is known about its effect on human lung cancer cells. The aim of the present study was to see whether Ph could induce apoptosis of non-small cell lung cancer (NSCLC) cells, and explore the possible underlying mechanism of action. We found that Ph markedly induced cell apoptosis of NSCLC cell line A549, and inhibited the migration of A549 cells in a dose-dependent manner. The expression level of BAX, cleaved caspase-3 and -9, and degraded form of PARP was increased and Bcl-2 was decreased after Ph treatment. In addition, the phosphorylation of P38 MAPK, ERK1/2 and JNK1/2 was increased in a dose‑dependent manner in parallel with Ph treatment. Inhibition of P38 MAPK and JNK1/2 by specific inhibitors significantly abolished the Ph-induced activation of the caspase-3 and -9. In vivo tumor-suppression assay further indicated that Ph (20 mg/kg) displayed a more significant inhibitory effect on A549 xenografts in tumor growth. All these findings indicate that Ph is able to inhibit NSCLC A549 cell growth by inducing apoptosis through P38 MAPK and JNK1/2 pathways, and therefore may prove to be an adjuvant to the treatment of NSCLC.
The role of NR4A1 in apoptosis is controversial. Pancreatic β-cells often face endoplasmic reticulum (ER) stress under adverse conditions such as high free fatty acid (FFA) concentrations and sustained hyperglycemia. Severe ER stress results in β-cell apoptosis. The aim of this study was to analyze the role of NR4A1 in ER stress-mediated β-cell apoptosis and to characterize the related mechanisms. We confirmed that upon treatment with the ER stress inducers thapsigargin (TG) or palmitic acid (PA), the mRNA and protein levels of NR4A1 rapidly increased in both MIN6 cells and mouse islets. NR4A1 overexpression in MIN6 cells conferred resistance to cell loss induced by TG or PA, as assessed by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay, and TUNEL assays indicated that NR4A1 overexpression also protected against ER stress-induced apoptosis. This conclusion was further confirmed by experiments exploiting siRNA to knockdown NR4A1 expression in MIN6 cells or exploiting NR4A1 knock-out mice. NR4A1 overexpression in MIN6 cells reduced C/EBP homologous protein (CHOP) expression and Caspase3 activation induced by TG or PA. NR4A1 overexpression in MIN6 cells or mouse islets resulted in Survivin up-regulation. A critical regulatory element was identified in Survivin promoter (-1872 bp to -1866 bp) with a putative NR4A1 binding site; ChIP assays demonstrated that NR4A1 physically associates with the Survivin promoter. In conclusion, NR4A1 protects pancreatic β-cells against ER stress-mediated apoptosis by up-regulating Survivin expression and down-regulating CHOP expression, which we termed as "positive and negative regulation."
Sodium butyrate (NaB) is a dietary microbial fermentation product of fiber and serves as an important neuromodulator in the central nervous system. In this study, we further investigated that NaB attenuated cerebral ischemia/reperfusion (I/R) injury in vivo and its possible mechanisms. NaB (5, 10 mg/kg) was administered intragastrically 3 h after the onset of reperfusion in bilateral common carotid artery occlusion (BCCAO) mice. After 24 h of reperfusion, neurological deficits scores were estimated. Morphological examination was performed by electron microscopy and hematoxylin-eosin (H&E) staining. The levels of oxidative stress and inflammatory cytokines were assessed. Apoptotic neurons were measured by TUNEL; apoptosis-related protein caspase-3, Bcl-2, Bax, the phosphorylation Akt (p-Akt), and BDNF were assayed by western blot and immunohistochemistry. The results showed that 10 mg/kg NaB treatment significantly ameliorated neurological deficit and histopathology changes in cerebral I/R injury. Moreover, 10 mg/kg NaB treatment markedly restored the levels of MDA, SOD, IL-1β, TNF-α, and IL-8. 10 mg/kg NaB treatment also remarkably inhibited the apoptosis, decreasing the levels of caspase-3 and Bax and increasing the levels of Bcl-2, p-Akt, and BDNF. This study suggested that NaB exerts neuroprotective effects on cerebral I/R injury by antioxidant, anti-inflammatory, and antiapoptotic properties and BDNF-PI3K/Akt pathway is involved in antiapoptotic effect.
In human hepatocellular carcinoma (HCC), aberrant expression of miRNAs correlates with tumor cell proliferation, apoptosis, invasion, and migration by targeting downstream proteins. miR-15b levels are reported increased in HCC tissues; however, they negatively correlate to HCC recurrence. Our aim was to understand the reason for this phenomenon. We used the reverse transcription-polymerase chain reaction (RT-PCR) to measure miR-15b-5p expression in both HCC tissues and HCC cell lines. Our results were consistent with the report. Using bioinformatics and luciferase reporter assays, we identified Rab1A as a novel and direct target of miR-15b-5p. Inhibiting the function of Rab1A with shRab1A also inhibited the growth of HCC cells and induced endoplasmic reticulum stress (ERS) and apoptosis. Similarly, suppressing Rab1A by overexpression of miR-15b-5p also inhibited cell growth and induced ERS and apoptosis. Moreover, re-expression of Rab1A rescued the miR-15b-5p-induced ERS, apoptosis, and growth inhibition in HCC cells. In vivo assays were further performed to testify them. Taken together, our data suggest that miR-15b-5p induces ERS, apoptosis, and growth inhibition by targeting and suppressing Rab1A, acting as a tumor suppressor gene in HCC. This finding suggests a novel relation among Rabs, miRNAs, and apoptosis.
We reported recently that after a nutritional growth retardation, rats showed significant weight gain, central fat accumulation, dyslipidemia, and β-cell dysfunction during a catch-up growth (CUG) phase. Here, we investigated whether glucagon-like peptide-1 (GLP-1) ameliorated the rapid weight gain, central fat deposition, and β-cell dysfunction during the CUG in rats. Sixty-four male Sprague Dawley rats were stratified into four groups including normal control group, CUG group, catch-up growth with liraglutide treatment group, and catch-up growth with liraglutide and exendin 9-39 treatment group. Energy intake, body weight, and body length were monitored. Fat mass percentage was analyzed by dual energy X-ray absorptiometry scan. Plasma triglyceride and non-esterified fatty acid were measured. The β-cell mass was analyzed by morphometric analysis and signaling molecules were examined by Western blot and real-time PCR. Insulin secretion capability was evaluated by hyperglycemic clamp test. Liraglutide prevented weight gain and improved lipid and glucose metabolism in rats under CUG conditions, which were associated with reduced fasting insulin levels and improved glucose-stimulated insulin secretion. Improved β-cell function is found to be associated with increased β-cell replication as determined by β-cell density and insulin-Ki67 dual staining. Furthermore, liraglutide increased islet pancreatic duodenal homeobox-1 (Pdx-1) and B-cell lymphoma-2 transcript and protein expression, and reduced Procaspase-3 transcript and Caspase-3 p11 subunit protein expression, suggesting that expression of Pdx-1 and reduction of apoptosis may be the mechanisms involved. The therapeutic effects were attenuated in rats co-administered with exendin 9-39, suggesting a GLP-1 receptor-dependent mechanism. These studies revealed that incretin therapy effectively prevented fast weight gain and β-cell dysfunction in rats under conditions of nutrition restriction followed by nutrition excess, which is in part due to enhanced functional β-cell mass and insulin secretory capacity.
Steroid alkaloids have been suggested as potential anticancer compounds. However, the underlying mechanisms of how steroid alkaloids inhibit the tumor growth are largely unknown. Here, we reported that solanine, a substance of steroid alkaloids, has a positive effect on the inhibition of pancreatic cancer cell growth in vitro and in vivo. In pancreatic cancer cells and nu/nu nude mice model, we found that solanine inhibited cancer cells growth through caspase-3 dependent mitochondrial apoptosis. Mechanically, solanine promotes the opening of mitochondrial membrane permeability transition pore (MPTP) by downregulating the Bcl-2/Bax ratio; thereafter, Cytochrome c and Smac are released from mitochondria into cytosol to process the caspase-3 zymogen into an activated form. Moreover, we found that the expression of tumor metastasis related proteins, MMP-2 and MMP-9, was also decreased in the cells treated with solanine. Therefore, our results suggested that solanine was an effective compound for the treatment of pancreatic cancer.
Emerging evidence indicate that mesenchymal stem cells (MSCs) affect tumor progression by reshaping the tumor microenvironment. Neutrophils are essential component of the tumor microenvironment and are critically involved in cancer progression. Whether the phenotype and function of neutrophils is influenced by MSCs is not well understood. Herein, we investigated the interaction between neutrophils and gastric cancer-derived MSCs (GC-MSCs) and explored the biological role of this interaction. We found that GC-MSCs induced the chemotaxis of neutrophils and protected them from spontaneous apoptosis. Neutrophils were activated by the conditioned medium from GC-MSCs with increased expression of IL-8, TNFα, CCL2, and oncostatin M (OSM). GC-MSCs-primed neutrophils augmented the migration of gastric cancer cells in a cell contact-dependent manner but had minimal effect on gastric cancer cell proliferation. In addition, GC-MSCs-primed neutrophils prompted endothelial cells to form tube-like structure in vitro. We demonstrated that GC-MSCs stimulated the activation of STAT3 and ERK1/2 pathways in neutrophils, which was essential for the functions of activated neutrophils. We further revealed that GC-MSCs-derived IL-6 was responsible for the protection and activation of neutrophils. In turn, GC-MSCs-primed neutrophils induced the differentiation of normal MSCs into cancer-associated fibroblasts (CAFs). Collectively, our results suggest that GC-MSCs regulate the chemotaxis, survival, activation, and function of neutrophils in gastric cancer via an IL-6-STAT3-ERK1/2 signaling cascade. The reciprocal interaction between GC-MSCs and neutrophils presents a novel mechanism for the role of MSCs in remodeling cancer niche and provides a potential target for gastric cancer therapy.
Musk has been traditionally used in East Asia to alleviate the symptoms of angina pectoris. However, it remains unclear as to whether muscone, the main active ingredient of musk, has any beneficial effects on persistent myocardial ischemia in vivo. The aim of the present study was to investigate whether muscone can improve cardiac function and attenuate myocardial remodeling following myocardial infarction (MI) in mice. Mice were subjected to permanent ligation of the left anterior descending coronary artery to induce MI, and then randomly treated with muscone (2 mg/kg/day) or the vehicle (normal saline) for 3 weeks. Sham-operated mice were used as controls and were also administered the vehicle (normal saline). Treatment with muscone significantly improved cardiac function and exercise tolerance, as evidenced by the decrease in the left ventricular end-systolic diameter, left ventricular end-diastolic diameter, as well as an increase in the left ventricular ejection fraction, left ventricular fractional shortening and time to exhaustion during swimming. Pathological and morphological assessments indicated that treatment with muscone alleviated myocardial fibrosis, collagen deposition and improved the heart weight/body weight ratio. Muscone inhibited the inflammatory response by reducing the expression of transforming growth factor (TGF)‑β1, tumor necrosis factor (TNF)-α, interleukin (IL)-1β and nuclear factor (NF)-κB. Treatment with muscone also reduced myocardial apoptosis by enhancing Bcl-2 and suppressing Bax expression. Muscone also induced the phosphorylation of protein kinase B (Akt) and endothelial nitric oxide synthase (eNOS). Our results demonstrate that muscone ameliorates cardiac remodeling and dysfunction induced by MI by exerting anti-fibrotic, anti-inflammatory and anti-apoptotic effects in the ischemic myocardium.
It has been documented in in vitro studies that zinc oxide nanoparticles (ZnO NPs) are capable of inducing oxidative stress, which plays a crucial role in ZnO NP-mediated apoptosis. However, the underlying molecular mechanism of apoptosis in neurocytes induced by ZnO NP exposure was not fully elucidated. In this study, we investigated the potential mechanisms of apoptosis provoked by ZnO NPs in cultured primary astrocytes by exploring the molecular signaling pathways triggered after ZnO NP exposure. ZnO NP exposure was found to reduce cell viability in MTT assays, increase lactate dehydrogenase (LDH) release, stimulate intracellular reactive oxygen species (ROS) generation, and elicit caspase-3 activation in a dose- and time-dependent manner. Apoptosis occurred after ZnO NP exposure as evidenced by nuclear condensation and poly(ADP-ribose) polymerase-1 (PARP) cleavage. A decrease in mitochondrial membrane potential (MMP) with a concomitant increase in the expression of Bax/Bcl-2 ratio suggested that the mitochondria also mediated the pathway involved in ZnO NP-induced apoptosis. In addition, exposure of the cultured cells to ZnO NPs led to phosphorylation of c-Jun N-terminal kinase (JNK), extracellular signal-related kinase (ERK), and p38 mitogen-activated protein kinase (p38 MAPK). Moreover, JNK inhibitor (SP600125) significantly reduced ZnO NP-induced cleaved PARP and cleaved caspase-3 expression, but not ERK inhibitor (U0126) or p38 MAPK inhibitor (SB203580), indicating that JNK signaling pathway is involved in ZnO NP-induced apoptosis in primary astrocytes.
The present study aims to investigate the pharmacological effect of the exopolysaccharides from Aphanothece halophytica GR02 (EPSAH) on the HeLa human cervical cancer cell line. HeLa cells were cultured in RPMI-1640-10% FBS medium containing with or without different concentrations of EPSAH. Cell viability was assessed by methylthiazol tetrazolium (MTT) assay. Cell apoptosis was elevated with Wright-Giemsa staining, AO/EB double staining, and DNA fragmentation assay. Apoptosis-associated molecules from cultured HeLa cells were quantified using Western blot analysis. Our results suggest that EPASH induces apoptosis in HeLa cells by targeting a master unfolded protein response (UPR) regulator Grp78. Grp78 further promotes the expression of CHOP and downregulates expression of survivin, which leads to activate mitochondria-mediated downstream molecules and p53-survivin pathway, resulting in caspase-3 activation and causing apoptosis. These findings provide important clues for further evaluating the potential potency of EPSAH for use in cancer therapy.
The fruit of Schisandra chinensis has been used in the traditional Chinese medicine for thousands of years. Accumulating evidence suggests that Schisandrin B (Sch B) has cardioprotection effect on myocardial ischemia in vitro. However, it is unclear whether Sch B has beneficial effects on continuous myocardial ischemia in vivo. The aim of the present study was to investigate whether Sch B could improve cardiac function and attenuate myocardial remodeling after myocardial infarction (MI) in mice. Mice model of MI was established by permanent ligation of the left anterior descending (LAD) coronary artery. Then the MI mice were randomly treated with Sch B or vehicle alone. After treatment for 3 weeks, Sch B could increase survival rate, improve heart function and decrease infarct size compared with vehicle. Moreover, Sch B could down-regulate some inflammatory cytokines, activate eNOS pathway, inhibit cell apoptosis, and enhance cell proliferation. Further in vitro study on H9c2 cells showed similar effects of Sch B on prevention of hypoxia-induced inflammation and cell apoptosis. Taken together, our results demonstrate that Sch B can reduce inflammation, inhibit apoptosis, and improve cardiac function after ischemic injury. It represents a potential novel therapeutic approach for treatment of ischemic heart disease.
ZFX (zinc finger transcription factor, X chromosome-linked) contributes to the maintenance of different types of stem cells and the progression of various cancers. We have previously reported that ZFX knockdown inhibits proliferation of glioma in vitro and in vivo. Since overexpression of ZFX in lung cancer tissue correlates with lymph node metastasis, we hypothesized that ZFX may play a role in lung cancer. In this study, we identified ZFX as a promoter of lung cancer growth and migration in a NSCLC (non-small cell lung carcinoma) cell line H1299. ZFX knockdown caused proliferation inhibition determined by MTT assay and colony formation assay, G0/G1 arrest of cell cycle and slightly increased proportion of apoptotic cells assessed by flow cytometry assay, decreased population of migrating cells showed by wound-healing assay, increased cell senescence evidenced by senescence-associated β-galactosidase staining. ZFX knockdown also led to decreased proportion of tumor bearing mice and reduced mean tumor volume in a subcutaneous tumor model. In addition, western blot showed that ZFX knockdown down regulated a set of proteins involved in proliferation, survival and motility. Altogether, these results suggest that ZFX may be a potential therapeutic target for NSCLC.
OBJECTIVE:
Alpinetin is a novel flavonoid that has demonstrated potent antitumor activity in previous studies. However, the efficacy and mechanism of alpinetin in treating lung cancer have not been determined.
METHODS:
We evaluated the impact of different doses and durations of alpinetin treatment on the cell proliferation, the apoptosis of lung cancer cells, as well as the drug-resistant lung cancer cells.
RESULTS:
This study showed that the alpinetin inhibited the cell proliferation, enhanced the apoptosis, and inhibited the PI3K/Akt signaling in lung cancer cells. Moreover, alpinetin significantly increased the sensitivity of drug-resistant lung cancer cells to the chemotherapeutic effect of cis-diammined dichloridoplatium. Taken together, this study demonstrated that alpinetin significantly suppressed the development of human lung cancer possibly by influencing mitochondria and the PI3K/Akt signaling pathway and sensitized drug-resistant lung cancer cells.
CONCLUSION:
Alpinetin may be used as a potential compound for combinatorial therapy or as a complement to other chemotherapeutic agents when multiple lines of treatments have failed to reduce lung cancer.
BACKGROUND:
Yi Qi Qing Re Gao (YQQRG) formula is a traditional Chinese herbal medicine used to treat chronic nephritis. This study was designed to evaluate the underlying mechanism in the use of YQQRG formula to treat nephrosis induced by puromycin aminonucleoside (PAN).
METHODS:
Thirty-six male Wistar rats were randomly divided into 3 groups of 12 rats each: a sham group, a vehicle-treated PAN model group (PAN), and a group treated with YQQRG (PAN + YQQRG). The PAN model was established by a single intravenous injection of PAN at a dose of 40 mg/kg body weight; rats in the sham group received the same volume of saline. Twenty-four hour urinary protein was measured 0, 3, 5, 10, and 15 days after the injection. The rats were sacrificed on day 10 and day 15 and the serum lipid profile examined. The renal cortex of each rat was stained with periodic acid-Schiff reagent and the pathologic alterations and ultrastructural changes were examined by transmission electron microscopy. In situ cell apoptosis was detected by a terminal deoxynucleotidyl transferase-mediated uridine 5'-triphosphate-biotin nick end-labeling (TUNEL) assay. Transcriptive levels of inflammatory markers and molecules associated with apoptosis were detected by a real-time polymerase chain reaction and expression of proteins was examined by either immunohistochemistry or Western blot analysis.
RESULTS:
YQQRG significantly decreased urinary protein level, and lowered serum lipid level. YQQRG also attenuated histologic lesions in the rat kidneys. Activation of inflammatory markers was largely restored by the administration of YQQRG. TUNEL assay showed that YQQRG decreased the number of apoptotic cells. Both mRNA and protein levels of caspase-3 were significantly reduced in the group treated with YQQRG, whereas expression of the Bcl-2 protein increased in the YQQRG group.
CONCLUSIONS:
YQQRG alleviated kidney injury in PAN-treated rats, possibly through anti-inflammatory and anti-apoptotic effects.
BACKGROUND:
Natural products from plants have been proven to be important resources of antitumor agents. In this study, we exploited the antitumor activity of (E)-3-(4-chlorophenyl)-N-(7-hydroxy-6-methoxy-2-oxo-2H-chromen-3-yl) acrylamide (SC-III3), a newly synthesized derivative of scopoletin, by in vitro and in vivo experiments.
METHODS:
Human hepatocellular carcinoma cell line HepG2 cells and xenograft of HepG2 cells in BALB/c nude mice were used to investigate the effects of SC-III3 on hepatocellular cancers. Cell cycle arrest and apoptosis were analyzed by flow cytometry. Cell cycle arrest, apoptosis and ATM-Chk pathway-related proteins were characterized by western blot.
RESULTS:
SC-III3 selectively inhibited the viability of HepG2 cells without significant cytotoxicity against human normal liver cells LO2. In mouse xenograft model of HepG2 cells, SC-III3 showed a marked inhibition of tumor growth in a dose-dependent manner. Cell cycle analysis revealed that SC-III3 induced cells to accumulate in S phase, which was accompanied by a marked decrease of the expressions of cyclin A, cyclin B, cyclin E and Cdk2 proteins, the crucial regulators of S phase cell cycle. SC-III3 treatment resulted in DNA breaks in HepG2 cells, which might contribute to its S phase arrest. The S arrest and the activation of ATM-Chk1/Chk2-Cdc25A-Cdk2 pathways induced by SC-III3 in HepG2 cells could be efficiently abrogated by pretreatments of either Ku55933 (an inhibitor of ATM) or UCN-01 (an inhibitor of Chk1/Chk2). The activation of p53-p21 pathway by SC-III3 was also reversed by Ku55933 treatment. SC-III3 led to significant accumulation of intracellular reactive oxygen species (ROS), a breaker of DNA strand, in HepG2 cells but not LO2 cells. Pretreatment with N-acetyl-l-cysteine (NAC), a ROS scavenger, could reverse SC-III3-caused ROS accumulation, DNA damage, activation of signal pathways relevant to DNA damage, S phase arrest and cell viability decrease in HepG2 cells.
CONCLUSION:
SC-III3 is able to efficiently inhibit the growth of hepatocellular carcinoma through inducing the generation of intracellular ROS, DNA damage and consequent S phase arrest, but lack of significant cytotoxicity against normal liver cells. This compound deserves further studies as a candidate of anticancer drugs.
BACKGROUND:
MicroRNAs (miRNAs) are frequently dysregulated in human cancers and can act as either potent oncogenes or tumor suppressor genes. In the present study, we intend to prove that the gene PTEN (phosphatase and tensin homolog deleted on chromosome ten) is a target gene of miR-205 and to investigate the suppressive effects on PTEN transcriptional activity by enhancing miR-205 expression in endometrial cancer Ishikawa cells.
METHODS:
Using Ishikawa cells as model systems, we up-regulated miR-205 expression by transient transfection with miR-205 mimics. A luciferase reporter assay, qRT-PCR and western blotting assays were used to verify whether PTEN is a direct target of miR-205. Meanwhile, the modulatory role of miR-205 in the AKT (protein kinase B) pathway was evaluated by determining the AKT phosphorylation. As a biological counterpart, we investigated cell apoptosis using flow cytometry.
RESULTS:
Our data indicate that miR-205 down-regulates the expression of PTEN through direct interaction with the putative binding site in the 3'-untranslated region (3'-UTR) of PTEN. Moreover, we documented the functional interactions of miR-205 and PTEN, which have a downstream effect on the regulation of the AKT pathway, explaining, at least in part, the inhibitory effects on Ishikawa cell apoptosis of enhancing miR-205 expression.
CONCLUSIONS:
For the first time, we demonstrate that the expression of PTEN is directly regulated by miR-205 in endometrial cancer cells and leads the inhibition of cellular apoptosis. This relationship could be targeted for new therapeutic strategies for endometrial cancer.
BACKGROUND AND PURPOSE:
Non-small cell lung cancer (NSCLC) is one of the most commonly diagnosed malignancies in the world. Small-molecule inhibitors of the EGF receptor's tyrosine kinase domain (TKIs), including gefitinib and erlotinib, have been widely used for treating NSCLC. Unfortunately, nearly all patients after initially experiencing a marked improvement while on these drugs, eventually progress to acquire resistance to TKIs. Because there is no effective therapeutic strategy to treat TKI-resistant NSCLC, we evaluated the effects of luteolin, a naturally occurring flavanoid, on T790M mutant NSCLC cells.
EXPERIMENTAL APPROACH:
The effect of luteolin on the viability of NSCLC and normal cell lines was investigated using the Cell Counting Kit-8 (CCK-8) assay. Luteolin-induced apoptosis was assessed by bivariate FITC-annexin V/PI assay, and Western blots were used to measured apoptotic proteins. Co-immunoprecipitation was used to determine the effect of luteolin on the interaction between Hsp90 and mutant EGF receptors. The effect of luteolin on the Akt/mTOR pathway was studied using Western blotting analysis. Its anti-tumour efficacy in vivo was examined in a mouse xenograft model.
KEY RESULTS:
Luteolin exerted significant anti-tumourigenic effects on the EGF receptor L858R/T790M mutation and erlotinib-resistant NSCLC both at the cellular and animal levels. Mechanistically, luteolin induced degradation of the EGF receptor by inhibiting the association of Hsp90 with the mutant EGF receptor, and, therefore, prevented PI3K/Akt/mTOR signalling, which resulted in NSCLC cell apoptosis.
CONCLUSION AND IMPLICATIONS:
Luteolin may be a potential candidate for NSCLC therapy, especially for treatment of patients with acquired erlotinib-resistant NSCLC.
© 2014 The British Pharmacological Society.
BACKGROUND:
To investigate the expression levels of importin13 (IPO13), c-kit, CD146, telomerase, caspase-3, bcl-2 and bax in endometrial polyps (EPs).
MATERIAL/METHODS:
We detected the mRNA expression levels of IPO13, c-kit, bcl-2 and bax in endometrial polyps (EPs) using real-time PCR. We detected the protein expression levels of IPO13, telomerase, CD146, caspase-3, bcl-2 and bax in EPs using S-P (Streptavidin-Peroxidase) immunohistochemistry. Western blotting was performed to determine the levels of importin13 and bcl-2 proteins in EPs.
RESULTS:
The expression levels of IPO13, c-kit, telomerase, caspase3, and bax were lower in the EP tissue compared to normal endometrial tissue during the proliferation and secretion phases of the menstrual cycle (p<0.05). The expression of CD146 was decreased in the EP tissue compared to the normal endometrial tissue during the proliferation phase of the menstrual cycle (p<0.05). The expression of bcl-2 was increased in the EP tissue compared to the normal endometrial tissue during the proliferation and secretion phases of the menstrual cycle (p<0.05).
CONCLUSIONS:
The expression levels of IPO13, c-kit, telomerase, caspase3, and bax were decreased; however, the expression of bcl-2 was increased in the EP tissue compared to the normal endometrial tissue. These findings suggest that the development of EPs is associated with the deregulated activities of the endometrial stem/progenitor cells and the decreased apoptosis of endometrial cells, with the latter being the major factor involved in the development of EPs.
The YiQiFuMai powder injection (YQFM), a traditional Chinese medicine (TCM) prescription re-developed based on the well-known TCM formula Sheng-maisan, showed a wide range of pharmacological activities in cardiovascular diseases in clinics. However, its role in protection against myocardial ischemia/reperfusion (MI/R) injury has not been elucidated. The present study not only evaluated the cardioprotective effect of YQFM from MI/R injury but also investigated the potential molecular mechanisms both in vivo and in vitro. The myocardium infarct size, production of lactate dehydrogenase (LDH), creatine kinase (CK), cardiac function, TUNEL staining, and caspase-3 activity were measured. Cell viability was determined, and cell apoptosis was measured by Hoechst 33342 staining and flow cytometry. Mitochondrial membrane potential (ΔΨm) was measured, and ATP content was quantified by bioluminescent assay. Expression of apoptosis-related proteins, including Caspase-3, Bcl-2, Bax, AMPKα, and phospho-AMPKα, was analyzed by western blotting. AMPKα siRNA transfection was also applied to the mechanism elucidation. YQFM at a concentration of 1.06 g/kg significantly reduced myocardium infarct size and the production of LDH, CK in serum, improved the cardiac function, and also produced a significant decrease of apoptotic index. Further, combined treatment with compound C partly attenuated the anti-apoptotic effect of YQFM. In addition, pretreatment with YQFM ranging from 25 to 400 μg/mL markedly improved cell viability and decreased LDH release. Moreover, YQFM inhibited H9c2 apoptosis, blocked the expression of caspase-3, and modulated Bcl-2 and Bax proteins, leading to an increased mitochondrial membrane potential and cellular ATP content. Mechanistically, YQFM activated AMP-activated protein kinase (AMPK) signaling pathways whereas pretreatment with AMPK inhibitor Compound C and application of transfection with AMPKα siRNA attenuated the anti-apoptotic effect of YQFM. Our results indicated that YQFM could provide significant cardioprotection against MI/R injury, and potential mechanisms might suppress cardiomyocytes apoptosis, at least in part, through activating the AMPK signaling pathways.
Lung cancer, especially non-small-cell lung cancer (NSCLC), plays the leading role in cancer which is closely related to a myriad of fatal results. Unfortunately, current molecular mechanisms and clinical treatment of NSCLC still remain to be explored despite the fact that intensive investigations have been carried out in the last two decades. Recently, growing attention to finding exploitable sources of anticancer agents is refocused on quinolone compounds, an antibiotic with a long period of clinic application, for their remarkable cell-killing activity against not only bacteria, but eukaryotes as well. In this study, we found LZ-106, an analog of enoxacin, exhibiting potent inhibitory effects on NSCLC in both cultured cells and xenograft mouse model. We identified apoptosis-inducing action of LZ-106 in NSCLC cells through the mitochondrial and endoplasmic reticulum (ER)-stress apoptotic pathways via Annexin-V/PI double-staining assay, membrane potential detection, calcium level detection and the expression analysis of the key apoptotic proteins. Through comet assay, reactive oxygen species (ROS) detection, the expression analysis of DNA damage response (DDR) marker γ-H2AX and other DDR-related proteins, we also demonstrated that LZ-106 notably induced ROS overproduction and DDR. Interestingly, additional evidence in our findings revealed that DDR and apoptosis could be alleviated in the presence of ROS scavenger N-acetyl-cysteine (NAC), indicating ROS-dependent DDR involvement in LZ-106-induced apoptosis. Thus our data not only offered a new therapeutic candidate for NSCLC, but also put new insights into the pharmacological research of quinolones.
Zinc finger protein, X-linked (ZFX) mediates the development and progression of human cancers. However, its potential role in chronic myeloid leukemia (CML) is still unknown. The ZFX expression was significantly increased in CML patients and cell lines. Based on loss-of-function experiments in CML cells, we found that knockdown of ZFX expression impaired cell proliferation and induced mitotic arrest in G0/G1 stage and apoptosis. In addition, ZFX silencing sensitized CML cells to imatinib treatment. Further, phospho-Akt (p-Akt), CyclinD1, CyclinE1, and Bcl-2 were downregulated, and Caspase-3 was upregulated in ZFX-silenced cells. In summary, our data suggest that ZFX is a novel oncogene promoting cell proliferation and inducing imatinib resistance via PI3K/Akt signaling pathway. ZFX may represent a potential therapeutic target in CML.
Oleuropein (OLE) was found to have anti-inflammatory and anti-oxidant effects. The latest study has shown that it can resist myocardial injury that follows an acute myocardial infarction and can rescue impaired spinal nerve cells. In this study, we investigated the neuroprotective effects of OLE on cerebral ischemia and reperfusion injury in a middle cerebral artery occlusion model in mice.OLE (100 mg/kg) was injected intraperitoneally 1h before ischemia. We found that the volume of cerebral infarction was significantly reduced after 75 min of ischemia and 24 h of reperfusion compared with the I/R (ischemia/reperfusion) group. This protective function occurred in a dose-dependent manner. We also found that treatment with OLE could reduce the cerebral infarct volume. The neuroprotective effect was prolonged from 2 h to 4 h when we injected OLE intracerebroventricularly after reperfusion. We then found that OLE can decrease the level of cleavedcaspase-3, an important marker of apoptosis, in the ischemic mouse brain. Finally, we explored the role of OLE in providing anti-apoptotic effects through the increased expression of Bcl-2 and the decreased expression of Bax, which are important markers in apoptosis. As shown above, the function and safety of OLE in cardiovascular disease may indicate that it is a potential therapeutic for stroke.
Cyclin D2 is involved in the pathology of vascular complications of type 2 diabetes mellitus (T2DM). This study investigated the role of cyclin-D2-regulated miRNAs in endothelial cell proliferation of T2DM. Results showed that higher glucose concentration (4.5 g/l) significantly promoted the proliferation of rat aortic endothelial cells (RAOECs), and significantly increased the expression of cyclin D2 and phosphorylation of retinoblastoma 1 (p-RB1) in RAOECs compared with those under low glucose concentration. The cyclin D2-3' untranslated region is targeted by miR-98, as demonstrated by miRNA analysis software. Western blot also confirmed that cyclin D2 and p-RB1 expression was regulated by miR-98. The results indicated that miR-98 treatment can induce RAOEC apoptosis. The suppression of RAOEC growth by miR-98 might be related to regulation of Bcl-2, Bax and Caspase 9 expression. Furthermore, the expression levels of miR-98 decreased in 4.5 g/l glucose-treated cells compared with those treated by low glucose concentration. Similarly, the expression of miR-98 significantly decreased in aortas of established streptozotocin (STZ)-induced diabetic rat model compared with that in control rats; but cyclin D2 and p-RB1 levels remarkably increased in aortas of STZ-induced diabetic rats compared with those in healthy control rats. In conclusion, this study demonstrated that high glucose concentration induces cyclin D2 up-regulation and miR-98 down-regulation in the RAOECs. By regulating cyclin D2, miR-98 can inhibit human endothelial cell growth, thereby providing novel therapeutic targets for vascular complication of T2DM.
Inflammation is emerging as a new hallmark of cancer. Arachidonic acid (AA) metabolism, the family of cyclooxygenases (COXs) and lipoxygenase (LOX) play important roles in AA-related inflammatory cascades. In 94 colorectal cancer samples collected from the Han population, the immunohistochemical results indicated that 68% of the patients with colorectal cancer had a co-expression of both COX-2 and 5-LOX, while both displayed low expression in the matched normal tissues. In cell lines, three colorectal cancer cell lines exhibited high expression of COX-2 and 5-LOX. During stable silencing of the expression of COX-2 or 5-LOX in LoVo cancer cells, we found that downregulation of either COX-2 or 5-LOX significantly diminished the growth, migration and invasion of the colon cancer cells and specifically, downregulation of COX-2 could elicit upregulation of 5-LOX protein and vice versa. The above results suggested that the simultaneous blocking of COX-2 and 5-LOX activity may bring more potential benefits in managing the progression of colon cancer. Therefore, we sought to explore the effectiveness of a dual COX-2/5-LOX inhibitor darbufelone on the proliferation, migration, invasion and apoptosis of colon cancer cells, as well as the underlying mechanism of action. The results indicated that darbufelone significantly decreased the proliferative and invasive abilities of the colon cancer cells, in a dose-dependent manner. During the study of the related mechanisms, we found an upregulation of p27 and downregulation of cyclin D1 as well as CDK4 after darbufelone treatment, which indicated that darbufelone could arrest the cell cycle of LoVo cells at the G0/G1 phase. Furthermore, the activation of caspase-3 and -9, upregulation of Bax and downregulation of Bcl-2 demonstrated the occurrence of apoptosis by darbufelone. Finally, darbufelone also prevented the migration and invasion of LoVo cells, which may be ascribed to the upregulation of E-cadherin and ZO-1. In summary, our data suggest that the inhibition of both COX-2/5-LOX may be an effective therapeutic approach for colon cancer management, particularly for those patients with high expression of COX-2/5-LOX.
Apelin has been proved to be protective against apoptosis induced by ischemic reperfusion. However, mechanisms whereby apelin produces neuroprotection remain to be elucidated. AMP-activated protein kinase (AMPK) is a master energy sensor that monitors levels of key energy metabolites. It is activated via AMPKαThr172 phosphorylation during cerebral ischemia and appears to be neuroprotective. In this study, we investigated the effect of apelin on AMPKα and tested whether apelin protecting against apoptosis was associated with AMPK signals. Focal transient cerebral ischemia/reperfusion (I/R) model in male ICR mice was induced by 60 min of ischemia followed by reperfusion. Apelin-13 was injected intracerebroventricularly 15 min before reperfusion. AMPK inhibitor, compound C, was injected to mice intraperitoneally at the onset of ischemia. In experiment 1, the effect of apelin-13 on AMPKα was measured. In experiment 2, the relevance of AMPKα and apelin-13' effect on apoptosis was measured. Data showed that apelin-13 significantly increased AMPKα phosphorylation level after cerebral I/R. Apelin-13, with the co-administration of saline, reduced apoptosis cells, down-regulated Bax and cleaved-caspase3 and up-regulated Bcl2. However, with the co-administration of compound C, apelin-13 was inefficient in affecting apoptosis and Bax, Bcl2 and cleaved-caspase3. The study provided the evidence that apelin-13 up-regulated AMPKα phosphorylation level in cerebral ischemia insults and AMPK signals participated in the mechanism of apelin-mediated neuroprotection.
To reveal SUMOylation and the roles of Sentrin-specific proteases (SENP)s in microglial cells under Intermittent hypoxia (IH) condition would provide more intensive view of understanding the mechanisms of IH-induced central nervous system (CNS) damage. Hence, in the present study, we detected the expression levels of SENPs in microglial cells under IH and normoxia conditions via RT-PCR assay. We found that SENP1 was significantly down-regulated in cells exposure to IH. Subsequently, the effect of IH for the activation of microglia and the potential roles of SENP1 in the SENP1-overexpressing cell lines were investigated via Western blotting, RT-PCR and Griess assay. The present study demonstrated the apoptosis-inducing and activating role of IH on microglia. In addition, we revealed that the effect of IH on BV-2 including apoptosis, nitric oxide synthase (iNOS) expression and nitric oxide (NO) induction can be attenuated by SENP1 overexpression. The results of the present study are of both theoretical and therapeutic significance to explore the potential roles of SENP1 under IH condition and elucidated the mechanisms underlying microglial survival and activation.
Uncontrolled endoplasmic reticulum (ER) stress activates members of the NOD-like receptor family, which are involved in the pyrin domain containing 3 (NLRP3) inflammasome pathway. This pathway has been proposed to contribute to β-cell dysfunction and death. However, the connection between ER stress and NLRP3 inflammasome activation remains controversial. Here we generated Akita/KO (Ins2(+/C96Y); NLRP3(-/-)) mice by crossing Akita (Ins2(+/C96Y); NLRP3(+/+)) mice with NLRP3 KO (Ins2(+/+); NLRP3(-/-)) mice. We then compared the metabolic phenotypes of the different strains. Knockout of the NLRP3 inflammasome did not affect the onset or the severity of diabetes in Akita/KO mice at any point of the study. Histological observations of pancreatic islets supported these findings. Tunicamycin-exposed islets from NLRP3 KO mice exhibited similar levels of ER stress and apoptosis induction as islets from WT (Ins2(+/+); NLRP3(+/+)) mice. Furthermore, NLRP3 deletion did not prevent tunicamycin-mediated reduction of glucose-stimulated insulin secretion. In conclusion, deletion of the NLRP3 inflammasome did not protect against ER stress-induced diabetes development or β-cell damage, indicating that β cell death in Akita mice is not mediated via activation of the NLRP3 inflammasome.
Evidence has shown that the activation of the autophagy pathway after experimental subarachnoid hemorrhage (SAH) protects against neuronal damage. Tert-butylhydroquinone (tBHQ), a commonly used nuclear factor erythroid 2-related factor 2 (Nrf2) activator, was found to significantly enhance autophagy activation. The aim of this study was to explore the effect of tBHQ treatment on early stage brain injury at 24 h after SAH. The results showed that tBHQ treatment failed to stimulate an effective anti-oxidative effect at 24 h after the SAH operation, but succeeded in ameliorating early brain injury, including alleviated brain edema, BBB disruption, neuronal degeneration and neurological deficits. Further exploration found that tBHQ treatment significantly increased the expression of Beclin-1 and the ratio of microtubule-associated protein 1 light chain 3 (LC3)-II to LC3-I, suggesting that autophagy was enhanced after tBHQ treatment. Moreover, tBHQ treatment restored Bcl-2 and Bax expression and reduced caspase-3 cleavage, suggesting the protective effect of tBHQ treatment in ameliorating brain injury after SAH. Furthermore, tBHQ enhanced autophagy activation, decreased neuronal degeneration and improved the neurological score after SAH in Nrf2-deficient mice. Taken together, these findings suggest that tBHQ treatment exerts neuro-protective effects against EBI following SAH by enhancing Nrf2-independent autophagy. Therefore, tBHQ is a promising therapeutic agent against EBI following SAH.
In this study, the anticancer effect of a newly synthesized flavonoid FV-429, against human breast cancer MDA-MB-231 cells, and the underlying mechanisms were investigated. FV-429 triggered the apoptosis and simultaneously inhibited the glycolysis of MDA-MB-231 cells. Both the HK II activity and its level in mitochondria were significantly down regulated by FV-429. Moreover, FV-429 weakened the interaction between HKII and VDAC, stimulated the detachment of HK II from the mitochondria, and resulted in the opening of the mitochondrial permeability transition pores. Thus FV-429 induced the mitochondrial-mediated apoptosis, showing increased Bax/Bcl-2 ratio, loss of mitochondrial membrane potential (MMP) and activation of caspase-3 and -9, cytochrome c (Cyt c) release, and apoptosis inducing factor (AIF) transposition. Further research revealed that the phosphorylation of mitochondrial HKII via Akt was responsible for the dissociation of HKII and the decreased HKII activity induced by FV-429. Taken together, FV-429 inhibited the phosphorylation of HKII, down-regulated its activity, and stimulated the release of HKII from the mitochondria, resulting the inhibited glycolysis and mitochondrial-mediated apoptosis. The studies provide a molecular basis for the development of flavonoid compounds as novel anticancer agents for breast cancer.
© 2015 Wiley Periodicals, Inc.
Nasopharyngeal carcinoma (NPC) is primarily treated by chemoradiation. However, how to promote radiation sensitivity in NPC remains a challenge. Salinomycin is potentially useful for the treatment of cancer. This study aimed to explore the radiosensitivity of salinomycin on human nasopharyngeal carcinoma cell line CNE-2. CNE-2 were treated with salinomycin or irradiation, alone or in combination. The cytotoxicity effects of salinomycin were measured using CCK-8 assay. Clonogenic survival assay was used to evaluate the effects of salinomycin on the radiosensitivity of CNE-2. The changes of cell cycle distribution and apoptosis were assayed using flow cytometry. The expression of Caspase3/Bax/Bal-2 was detected by Western blotting. DNA damage was detected via γ-H2AX foci counting. The results showed that salinomycin induced apoptosis and G2/M arrest, increased Bax and cleaved Caspase3, decreased Bcl-2 expression, and increased the formation of γ-H2AX nuclear foci. These data suggest that salinomycin may be a radiosensitizer for NPC radiotherapy.
L-Theanine is an amino acid derivative from green tea. The present work was aimed at the effect of L-theanine on neuron-like rat pheochromocytoma (PC12) cells stimulated with cadmium chloride. Treatment with L-theanine before cadmium exposure increased cell viability; the experiments of Annexin V/PI staining indicated that L-theanine inhibited cadmium-induced cell apoptosis. Meanwhile, L-theanine decreased ROS production and protected from cadmium-induced disruption of mitochondrial transmembrane potential. Compared with cadmium-treated cells, L-theanine could also decrease the ratio of Bax/Bcl-2, as well as the level of cleaved caspase-9, caspase-3 and poly(ADP-ribose) polymerase. Furthermore, L-theanine depresses cadmium-induced up regulation of phosphorylations of PI3K/Akt, MAPK ERK1/2, and JNK signaling. These data suggest that L-theanine pretreatment reduces severity of cadmium toxicity probably via antioxidant action. Therefore, it may be concluded that L-theanine could be exploited for prevention of cadmium-induced diseases.
Adrenomedullin (ADM), a multifunctional regulatory peptide, is potentially induced by hypoxia in physiological and pathological tissues, including many types of malignant tumors. Recent research has demonstrated that ADM expression is highly associated with the prognosis and disease severity of human osteosarcoma. However, the effect of ADM on the apoptosis of osteosarcoma cells and its possible mechanism remain to be elucidated. In the present study, we observed that mRNA and protein levels of ADM were increased in human osteosarcoma SOSP-F5M2 cells under a hypoxic microenvironment induced by cobalt chloride (CoCl2) in a time-dependent manner. Treatment with ADM significantly blunted hypoxic-induced apoptosis, evaluated by Hoechst 33342 staining and Annexin V-FITC/PI labeling. The expression of B-cell lymphoma-2 (Bcl-2) was increased by administration of ADM; meanwhile, this effect was reversed by exogenously adding U0126, a selective inhibitor of MEK or ADM22-52 (ADM-specific receptor antagonist). These results demonstrated that ADM acted as a survival factor to inhibit hypoxic-induced apoptosis via interacting with its receptors CRLR-RAMP (2,3) in osteosarcoma cells. The anti-apoptotic function of ADM was found to be mediated by upregulation of the expression of Bcl-2 partially through activation of the MEK/ERK1/2 signaling pathway. Therefore, targeting of the ADM/ADM acceptors/ERK1/2/Bcl-2 pathway may provide a potential strategy through which to induce the apoptosis of osteosarcoma cells.
Lithium has been reported to have neuroprotective effects, but the preventive and treated role on cognition impairment and the underlying mechanisms have not been determined. In the present study, C57Bl/6 mice were subjected to repeated bilateral common carotid artery occlusion to induce the learning and memory deficits. 2 mmol/kg or 5 mmol/kg of lithium chloride (LiCl) was injected intraperitoneally per day before (for 7 days) or post (for 28 days) the operation. This repeated cerebral ischemia-reperfusion (IR) induced dynamic overexpression of ratio of Bcl-2/Bax and BDNF in hippocampus of mice. LiCl pretreatment and treatment significantly decreased the escape latency and increased the percentage of time that the mice spent in the target quadrant in Morris water maze. 2 mmol/kg LiCl evidently reversed the morphologic changes, up-regulated the survival neuron count and increased the BDNF gene and protein expression. 5 mmol/kg pre-LiCl significantly increased IR-stimulated reduce of Bcl-2/Bax and p-CREB/CREB. These results described suggest that pre-Li and Li treatment may induce a pronounced prevention on cognitive impairment. These effects may relay on the inhibition of apoptosis and increasing BDNF and p-CREB expression.
O(2)-(2,4-dinitro-5-{[2-(12-en-28-b-d-galactopyranosyl-oleanolate-3-yl)-oxy-2-oxoethyl]amino}phenyl)1-(N-hydroxyethylmethylamino)diazen-1-ium-1,2-diolate (NOAD), a novel NO-releasing derivative of oleanolic acid (OA), is an active cytotoxic component. In this study, NOAD induced a rise in intracellular NO levels and showed cytotoxic effects which were prevented by hemoglobin (NO scavenger). Meanwhile, NOAD induced G2/M phase cell cycle arrest in a concentration-dependent manner. Analysis of the cell cycle regulatory proteins demonstrated that NOAD did not change the steady-state levels of cyclin A, cyclin B, cyclin E, Cdk2 and Cdk4, but decreased the protein levels of Cdk1 and Cdc25C. Meanwhile, the levels of phosphorylation of Cdc25C and Cdk1 were significantly increased by NOAD in a concentration-dependent manner. Moreover, NOAD modulated the phosphorylation of protein kinases Chk2. During the G2/M arrest, cyclin-dependent kinase inhibitors (CDKIs), p21(WAF1/CIP1) and p27(kip1) were increased in a concentration-dependent manner. In addition, NOAD also caused a marked increase in the apoptotic cells, as characterized by fragmented nuclei, sub G1 formation, the level of 8-OHDG increase and poly (ADP-ribose) polymerase (PARP) cleavage, which was associated with activation of caspase-3, caspase-8 and caspase-9. Up-regulation of Bax and down-regulation of Bcl-2 were also observed in Bel-7402 cells treated with NOAD. These data suggest that NOAD produces anti-tumor effect via induction of G2/M cell cycle arrest and apoptosis.
Multiple sclerosis (MS) has been associated with a history of sub-optimal exposure to ultraviolet light, implicating vitamin D3 as a possible protective agent. We evaluated whether 1,25(OH)2D3 attenuates the progression of experimental autoimmune encephalomyelitis (EAE), and explored its potential mechanisms. EAE was induced in C57BL/6 mice via immunization with MOG35-55, and some mice received 1,25(OH)2D3. 1,25(OH)2D3 inhibited EAE progression. Additionally, 1,25(OH)2D3 reduced inflammation, demyelination, and neuron loss in the spinal cord. The protective effect of 1,25(OH)2D3 was associated with significantly elevated expression of Beclin1, increased Bcl-2/Bax ratio, and decreased LC3-II accumulation. Thus, 1,25(OH)2D3 may represent a promising new MS treatment.
Human non-small cell lung carcinoma (NSCLC) is one of the most common cancer worldwide. In previous studies, lovastatin, acting as an inhibitor of 3-hydroxy-3-methylglutaryl Co A (HMG-CoA) reductase, exhibited significant antitumor activity during tumorigenesis. However, whether or not this effect is mediated through changes in minichromosome maintenance (MCM) 2 expression remains unclear. The present study investigated whether lovastatin inhibits proliferation due to MCM2 in NSCLCs. We first assessed the effects of lovastatin on cell anti-proliferation, cell cycle progression and apoptosis in NSCLC cells. We found, by quantitative RT-PCR and western blot analysis, that lovastatin treatment markedly and consistently inhibited the expression of MCM2. Then, to further explore the anticancer mechanism of lovastatin involving MCM2, we silenced MCM2 by siRNA in two cell lines (A549 and GLC-82). Silencing of MCM2 triggered G1/S arrest. Following further examination of cell cycle-related factors, MCM2 knockdown inhibited protein retinoblastoma (Rb), cyclin D1 and CDK4 expression, but increased p21 and p53 expression, suggesting that siMCM2 indeed triggered cell cycle arrest. In addition, siMCM2 induced apoptosis. Finally, lovastatin treatment increased p-JNK, which is involved in the downregulation of MCM2. In conclusion, our data suggest that MCM2 may be a novel therapeutic target of lovastatin treatment in NSCLCs.
Ischemic stroke is the third leading cause of death and the main reason for severe disabilities in the world today. N2, 4 - (2 - (1H - imidazol - 1 - yl) ethoxy) - 3 - methoxybenzoic acid is considered as a novel potent agent for cerebral ischemia due to its effect in preventing neuronal cell death after ischemic stroke. In the present study, we investigated the post-ischemic neuroprotective effect of N2 and its underlying mechanisms. Using a MCAO rat model, we found that N2 reversed brain infarct size, reduced cerebral edema and decreased the neurological deficit score significantly. Moreover, N2 diminished TUNEL positive cells, down-regulated bax expression and up-regulated bcl-2 expression notably. In addition, we evaluated the oxygen glucose deprivation/reoxygenation (OGD/R) injury induced neuron cell death in rat primary cortical neuron and assessed the neuroprotective effect of our drug. N2 increased cell viability, ameliorated neuron cell injury by decreasing LDH activity, and inhibited cell apoptotic rate while suppressed apoptotic signaling via inhibiting the bax expression, and elevating the bcl-2 expression. Furthermore, the neuroprotective effect of N2 was associated with the PI3K/Akt pathway which was proved by the use of PI3K inhibitor LY294002. The combination of our findings disclosed that N2 can be used as an effective neuroprotective agent for ischemic stroke due to its significant effect on preventing neuronal cell death after cerebral ischemia both in vivo and in vitro and the effectiveness was dose dependent.
Survivin is an important protein in regulating both cell apoptosis and proliferation. It has attracted growing attentions in recent years as a promising target for cancer therapy. Previous studies have revealed that monomeric survivin regulated apoptosis in a more significant way than the wild-type survivin that generally contains a large portion of its dimers. In order to investigate the roles of monomeric mutant survivin apoptosis and cell cycle regulation of human cancer cells, we developed and tested three dominant-negative mutants with multisite mutations (MSM) including TAT-survivin(34/101/102), TAT-survivin(34/117/101/102) and TAT-survivin(117/101/102). Results revealed that MSM mutants remained as monomers under ambient conditions, and induced cells (breast cancer Bcap-37 cells) apoptosis even more efficiently, primarily through the caspase-dependent and Bcl-2-related pathways, than non-monomeric mutants. We further identified that the TAT-survivin(34/101/102) and TAT-survivin(117/101/102) MSM significantly inhibited the proliferation of Bcap-37 cells and arrested cells in S and G2/M phases, while TAT-survivin(34/117/101/102) arrested cells in G2/M phase. It appeared to us that TAT-survivin(34/101/102) and TAT-survivin(117/101/102) also inhibited cell proliferation more significantly. These findings suggest that such MSM afford monomeric survivin with promising potentials for cancer therapy.
Microenvironmental hypoxia gives many tumor cells the capacity for drug resistance. Thioredoxin family members play critical roles in the regulation of cellular redox homeostasis in a stressed environment. In this study, we established a hypoxia-drug resistance (hypoxia-DR) model using HepG2 cells and discovered that the overexpression and nuclear translocation of thioredoxin-1 (Trx-1) are closely associated with this resistance through the regulation of the metabolism by the oxidative stress response to glycolysis. Intranuclear Trx-1 enhances the DNA-binding activity of HIF-1α via its interaction with and reducing action on Ref-1, resulting in increased expression of glycolysis-related proteins (PDHK1, HKII, and LDHA), glucose uptake, and lactate generation under hypoxia. Meanwhile, we found that GL-V9, a newly synthesized flavonoid derivative, shows an ability to reverse the hypoxia-DR and has low toxicity both in vivo and in vitro. GL-V9 could inhibit the expression and nuclear translocation of Trx-1 and then suppress HIF-1α DNA-binding activity by inhibiting the Trx-1/Ref-1 axis. As a result, glycolysis is weakened and oxidative phosphorylation is enhanced. Thus, GL-V9 leads to an increment in intracellular ROS generation and consequently intensified apoptosis induced by DDP.
The dysregulation of miR-1224-5p has been reported in several human cancers. However, the expression and function of miR-1224-5p in glioma remains unknown. The aim of our study was to investigate the effect of miR-1224-5p on glioma cells and to determine its functional signaling mediators. Using 198 glioma samples within the Chinese Glioma Genome Atlas expression dataset, we demonstrated that miR-1224-5p expression is decreased in high-grade gliomas when compared with low-grade gliomas. Differential miR-1224-5p expression in 50 randomly selected samples was verified by in situ hybridization. The expression of miR-1224-5p was shown to positively correlate with overall survival in 82 glioblastoma patients. Exogenous expression of miR-1224-5p in glioma cells suppressed proliferation and invasion and promoted apoptosis. Target prediction algorithms identified a consensus miR-1224-5p recognition site in the 3'UTR of the cAMP response element-binding protein (CREB1) gene, and this sequence was shown to directly confer miR-1224-5p repression in luciferase reporter assays. Furthermore, exogenous miR-1224-5p expression was shown to down-regulate CREB1, as well as its downstream target genes matrix metalloproteinase-9 and B-cell lymphoma-2. Conversely, over-expression of CREB1 reversed the effect of miR-1224-5p on the proliferation, invasion, and apoptosis of glioma cells. These data indicate that miR-1224-5p may inhibit tumor-associated activity in malignant gliomas by targeting CREB1.
Recombinant hirudin (rH) is a highly potent and specific inhibitor of thrombin, and has been shown to inhibit the growth and metastasis of several types of cancers in experimental tumor models. The objective of this study was to evaluate the antitumor effects and explore the underlying mechanisms of rH in Hep-2 human laryngeal carcinoma (LC) cells. Hep-2 cells were treated with various concentrations of rH for 24 h. The cell viability was evaluated by a water-soluble tetrazolium salt (WST) assay. The adhesion ability of the cells was evaluated by cell adhesion to fibronectin. Cell migration and invasion were measured with the Boyden chamber assay. Cell apoptosis was detected by Hoechst 33324 fluorescence staining. A chicken chorioallantoic membrane (CAM) assay was used to assess the effects of rH on angiogenesis in vivo. Western blotting was used to detect the expression levels of vascular endothelial growth factor receptor (VEGF-R), focal adhesion kinase (FAK), Bcl-2-associated agonist of cell death (Bad) and B-cell CLL/lymphoma 2 (Bcl-2) proteins. rH significantly inhibited the cell viability and induced apoptosis in LC Hep-2 cells in a dose-dependent manner, as compared with phosphate-buffered saline (PBS) as control. These results were accompanied by a decrease in the anti-apoptotic protein Bcl-2 and an increase in the pro-apoptotic protein Bad. Moreover, rH dose-dependently inhibited the adhesion, migration and invasion of the Hep-2 cells, compared to the vehicle PBS. In addition, rH robustly suppressed angiogenesis in the CAM assay. Importantly, the expression of adhesion and angiogenesis-associated proteins FAK and VEGF-R was significantly downregulated by rH in a dose-dependent manner. The present findings demonstrate that rH exerts antitumor effects in Hep-2 human laryngeal cancer cells via multiple mechanisms and suggests that targeting thrombin by rH is a potential strategy for the treatment of LC.
During the past decades, many efforts have been made in mimicking the clinical progress of human cancer in mouse models. Previously, we developed a human breast tissue-derived (HB) mouse model. Theoretically, it may mimic the interactions between "species-specific" mammary microenvironment of human origin and human breast cancer cells. However, detailed evidences are absent. The present study (in vivo, cellular, and molecular experiments) was designed to explore the regulatory role of human mammary microenvironment in the progress of human breast cancer cells. Subcutaneous (SUB), mammary fat pad (MFP), and HB mouse models were developed for in vivo comparisons. Then, the orthotopic tumor masses from three different mouse models were collected for primary culture. Finally, the biology of primary cultured human breast cancer cells was compared by cellular and molecular experiments. Results of in vivo mouse models indicated that human breast cancer cells grew better in human mammary microenvironment. Cellular and molecular experiments confirmed that primary cultured human breast cancer cells from HB mouse model showed a better proliferative and anti-apoptotic biology than those from SUB to MFP mouse models. Meanwhile, primary cultured human breast cancer cells from HB mouse model also obtained the migratory and invasive biology for "species-specific" tissue metastasis to human tissues. Comprehensive analyses suggest that "species-specific" mammary microenvironment of human origin better regulates the biology of human breast cancer cells in our humanized mouse model of breast cancer, which is more consistent with the clinical progress of human breast cancer.
Angiogenin (ANG) is a multifunctional secreted protein that belongs to the pancreatic ribonuclease A super family, which has been conceived to play a more important role in cell survival, growth and proliferation than the mediation of angiogenesis. Accumulating evidences suggest that the expression and activity of ANG increased significantly in a variety of human cancers. Recent studies showed that ANG activates cell signaling pathway through the putative receptor on endothelial cells. However, the underlying mechanisms remain largely unknown. AKT/mTOR signaling pathway participates in cell growth, cell-cycle progression and cell apoptosis. The purpose of our study was to determine whether ANG implicated in growth and metastasis of bladder cancer cells through regulating AKT/mTOR signaling pathway. In this study, we constructed ANG siRNA plasmids that transfected into human bladder cancer T24 cells. We demonstrated that knockdown of ANG could inhibit cell proliferation, regulate cell cycle and induce apoptosis. We also found that down-regulation of ANG remarkably reduced the phosphorylation of signaling targets AKT, GSK-3β and mTOR. Furthermore, down-regulation of ANG increased expression of ribonuclease inhibitor, which is a cytoplasmic acidic protein with many functions. Finally, ANG siRNA led to the suppression for tumorigenesis and metastasis in vivo. Taken together, these findings highlight for the first time that ANG could play a pivotal role in the development of bladder cancer through regulating AKT/mTOR signaling pathway. The targeting of ANG and associated factors could provide a novel strategy to inhibit human bladder cancer.
Two myelodysplastic syndrome (MDS) cell lines, MUTZ-1 and SKM-1 cells, were used to study the effect of arsenic trioxide (As2O3) on hematological malignant cells. As2O3 induced this two cell lines apoptosis via activation of caspase-3/8 and cleavage of poly (ADP-ribose) polymerase (PARP), a DNA repair enzyme. As2O3 reduced NF-κB activity, which was important for inducing MUTZ-1 and SKM-1 cells apoptosis. As2O3 also inhibited the activities of hTERT in MUTZ-1 and SKM-1 cells. Moreover, the NF-κB inhibitor, pyrrolidine dithiocarbamate (PDTC), had no effect on caspase-8 activation, although PDTC did inhibit MUTZ-1 and SKM-1 cells proliferation. Incubation of MUTZ-1 cells with a caspase-8 inhibitor failed to block As2O3-induced inhibition of NF-κB activity. Our findings suggest that As2O3 may induce apoptosis in MUTZ-1 and SKM-1 cells by two independent pathways: first, by activation of caspase-3/8 and PARP; and second, by inhibition of NF-κB activity, which results in downregulation of hTERT expression. We conclude that hTERT and NF-κB are important molecular targets in As2O3-induced apoptosis.
NG, O(2)-(2,4-dinitro-5-{[2-(12-en-28-β-D-galactopyranosyl-oleanolate-3-yl)-oxy-2-oxoethyl] amino} phenyl) 1-(N-hydroxyethylmethylamino) diazen-1-ium-1,2-diolate, was identified in our laboratory as a novel nitric oxide-releasing prodrug with antitumor effects. A previous study showed that NG inhibited cell growth, and induced apoptosis in HepG2 cells. In this study, the inhibitory effects of NG on the viability of MGC803 cells were examined using methylthiazolyl tetrazolium biomide (MTT) assay, neutral red assay and trypan blue exclusion test. The results showed that NG had strong cytotoxicity to induce apoptosis, which was characterized by a significant externalization of phosphatidylserine, nuclear morphological changes and enhanced Bax-to-Bcl-2 ratio. Moreover, the release of cytochrome c (Cyt c) from mitochondria and the activation of caspase-9/3 were also detected, indicating that NG may induce apoptosis through a mitochondrial-mediated pathway. NG induced mitochondrial dysfunction in MGC803 cells by altering membrane potential (△Ψm), the inhibition of complexes I, II and IV consequently decreasing ATP level. Furthermore, the treatment of MGC803 cells with NG caused a marked rise in oxidative stress as characterized by accumulation of reactive oxygen species (ROS), excessive malondialdehyde (MDA) production and a reduction in glutathione hormone (GSH) level and superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px) activity. In addition, pretreatment with N-acetylcysteine (NAC), a GSH synthesis precursor, was partially protective against the NG-induced ROS generation and cell apoptosis. In contrast, pretreatment of MGC803 cells with L-buthionine-S, R-sulfoximine (BSO), a GSH synthesis inhibitor, increased the ROS levels, and aggravated cell apoptosis by NG. These results suggest that NG-induced apoptosis in MGC803 cells is mediated, at least in part, by the increase in ROS production, oxidative stress and mitochondrial dysfunction.
Stellate ganglion block (SGB) is a blockade of sympathetic ganglia innervating the head and neck, and is known to function through vasodilation of the target region. However, the effectiveness of SGB in relieving cerebral vasospasm (CVS) through dilation of intracerebral vessels has not been evaluated. The aim of the present study is to investigate the therapeutic effects of SGB in a rat model of subarachnoid hemorrhage (SAH) complicated by delayed CVS, and explore the underlying mechanisms. The SAH model was established by double injection of autologous arterial blood into the cisterna magna. We simulated SGB by transection of the cervical sympathetic trunk (TCST), and measured changes in the diameter, perimeter and cross-sectional area of the basilar artery (BA) and middle cerebral artery (MCA) to evaluate its vasodilatory effect. To investigate the underlying mechanisms, we determined the expression level of vasoactive molecules endothelin-1 (ET-1) and calcitonin gene-related peptide (CGRP) in the plasma, and apoptotic modulators Bcl-2 and Bax in the hippocampus. We found a significant increase in the diameter, perimeter and cross-sectional area of the BA and right MCA in SAH rats subjected to TCST. Application of SGB significantly reduced the expression of ET-1 while increasing that of CGRP in SAH rats. We also found a significant increase in the expression of Bcl-2 and decrease in the expression of Bax in the hippocampus of SAH rats subjected to TCST, when compared to untreated SAH rats. The mechanism of action of SGB is likely mediated through alterations in the ratio of ET-1 and CGRP, and Bax and Bcl-2. These results suggest that SGB can alleviate the severity of delayed CVS by inducing dilation of intracerebral blood vessels, and promoting anti-apoptotic signaling. Our findings provide evidence supporting the use of SGB as an effective and well-tolerated approach to the treatment of CVS in various clinical settings.
MicroRNAs are a class of noncoding small RNAs that regulate gene expression by inhibiting target genes at post-transcriptional levels. MicroRNAs have been highlighted in many organs and tissues, including the brain. To identify special microRNAs involved in ischemia-reperfusion injury, we performed a comprehensive small RNA profiling in rat model and the control using Illumina high-throughput sequencing. A total of 9,444,562 and 10,290,391 clean reads were sequenced from two small RNA libraries constructed, respectively. Three hundred fifty-eight known microRNAs were identified, in which 78 microRNAs exhibited significantly differential expression between model and control. In addition, 62 and 68 novel miRNAs were found in model and control, respectively. Comparative analysis showed that 24 novel microRNAs were differentially expressed with greater than six-fold change. The GO annotation suggested that predicted targets of microRNAs were enriched into the category of metabolic process, cell part, cell-extracellular communications, and so on. KEGG pathway analysis suggested that these genes were involved in many important pathways, mainly including signaling transduction, MAPK signaling pathway, NF-κB signaling pathway, and neurotrophin signaling pathway. Our findings provided a deeper understanding to the regulatory mechanism of microRNAs underlying cerebral ischemia, therefore benefitting the improvement of the protection and treatment strategies of this disease.
Human ribonuclease inhibitor (RI), a cytoplasmic protein, is constructed almost entirely of leucine rich repeats. RI could suppress activities of ribonuclease and angiogenin (ANG) through closely combining with them. ANG is a potent inducer of blood vessel growth and has been implicated in the establishment, growth, and metastasis of tumors. ILK/PI3K/AKT signaling pathway also plays important roles in cell growth, cell-cycle progression, tumor angiogenesis, and cell apoptosis. Our previous experiments demonstrated that RI might effectively inhibit some tumor growth and metastasis. Our recent study showed that ILK siRNA inhibited the growth and induced apoptosis in bladder cancer cells as well as increased RI expression, which suggest a correlation between RI and ILK. However, the exact molecular mechanism of RI in anti-tumor and in the cross-talk of ANG and ILK signaling pathway remains largely unknown. Here we investigated the effects of up-regulating RI on the growth and apoptosis in murine melanoma cells through angiogenin and ILK/PI3K/AKT signaling pathway. We demonstrated that up-regulating RI obviously decreased ANG expression and activity. We also discovered that RI overexpression could remarkably inhibit cell proliferation, regulate cell cycle and induce apoptosis. Furthermore, up-regulation of RI inhibited phosphorylation of ILK downstream signaling targets protein kinase B/Akt, glycogen synthase kinase 3-beta (GSK-3β), and reduced β-catenin expression in vivo and vitro. More importantly, RI significant inhibited the tumor growth and angiogenesis of tumor bearing C57BL/6 mice. In conclusion, our findings, for the first time, suggest that angiogenin and ILK signaling pathway plays a pivotal role in mediating the inhibitory effects of RI on melanoma cells growth. This study identifies that RI may be a useful molecular target for melanoma therapy.
Apelin has been proved to protect the heart against ischemia/reperfusion (I/R) injury via the activation of phosphatidylinositol 3-kinase (PI3K)/Akt and extracellular signal-regulated kinase (ERK) signaling pathways. Whether this protective effect applies to brain I/R injury needed to be explored. We therefore investigated the potential neuroprotective role of Apelin-13 and the underlying mechanisms. Focal transient cerebral I/R model in male ICR mice was induced by 60min of ischemia followed by reperfusion. Apelin-13 intracerebroventricular injection was performed 15 min before reperfusion. Neurological function, infarct volume, brain edema and apoptosis were measured at 24h after stroke. To further test the mechanism of Apelin-13, PI3K inhibitor LY294002 and ERK1/2 inhibitor PD98059 were injected into the lateral cerebral ventricle 15min before ischemia. Compared with the Vehicle group, Apelin-13 significantly ameliorated neurological deficit, infarct volume, brain edema and reduced TUNEL-positive cells. Bax, caspase-3 and cleaved caspase-3 were down-regulated and Bcl-2 up-regulated. While, the effect of Apelin-13 on Bax, Bcl-2, caspase-3 and cleaved caspase-3 was attenuated by LY294002 and PD98059. Apelin protected the brain against I/R insult injury, and this effect may be through activation of PI3K/Akt and ERK1/2 signaling pathways.
Millet is an important cereal food and exhibits multiple biological activities, including immunodulatory, antioxidant, antifungal and anti-hyperglycemia effects. Herein, we describe a novel 35kDa protein with anti-cancer properties, named FMBP, which was extracted and purified from foxtail millet bran by cell-based screening. FMBP is highly homologous to peroxidase as revealed by mass spectrometry and gene sequencing analysis. Furthermore, in vivo anti-tumor results implicated that the novel protein FMBP had the ability to suppress xenografted tumor growth in nude mice. Mechanistically, FMBP is able to suppress colon cancer cell growth through induction of G1 phase arrest and also causes a loss of mitochondrial transmembrane potential which results in caspase-dependent apoptosis in colon cancer cells. Notably, FMBP has much lower toxicity in normal colon epithelial cells. Taken together, FMBP has targeted anti-colon cancer effects and may serve as a therapeutic agent against colon cancer.
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by pronounced synovial inflammation and hyperplasia, in which there may be an imbalance between the growth and death of fibroblast-like synoviocytes (FLS). Norisoboldine (NOR), the main active constituent in the alkaloid fraction isolated from Radix Linderae, was previously demonstrated to alleviate arthritis severity in experimental RA. This study aimed to evaluate the effects of NOR on proliferation and apoptosis of FLS from adjuvant-induced arthritis (AIA) rats to elucidate the mechanism of its inhibitory effect on inflammatory synovial hyperplasia in RA. Our results indicated that NOR exhibited a pro-apoptotic effect on AIA FLS but only slightly affected cell proliferation and the cell cycle. Following treatment with NOR for 24h, the activation of caspase 3 and caspase 9 and the cleavage of poly (ADP-ribose) polymerase (PARP) in AIA FLS were observed; however, caspase 8 remained unaffected. Meanwhile, a flow cytometric assay revealed that NOR significantly increased the percentage of apoptotic cells, causing the loss of the depolarized mitochondrial membrane potential and the release of cytochrome C. The expression of Bax and Bcl-2 was also regulated by NOR treatment. Additionally, the expression of p53 protein was up-regulated by NOR, and pretreatment with PFT-α, a p53 specific inhibitor, reversed the increase in FLS apoptosis caused by NOR. These findings indicated that NOR-induced apoptosis in AIA FLS is achieved via a mitochondrial-dependent pathway, which may be mediated by promoting the release of cytochrome C and by regulating the expression of Bax and Bcl-2 proteins, and p53 might also be required for NOR-induced apoptosis in AIA FLS.
(S)-ZJM-289, a novel nitric oxide (NO)-releasing derivative of 3-n-butylphthalide, induces the neuroprotection in a rat model of focal cerebral ischemia/reperfusion (I/R). However, much is unknown about the late phase effect in the neuroprotection of (S)-ZJM-289 preconditioning. The purpose of this study is to explore the late phase neuroprotection of (S)-ZJM-289 preconditioning, as well as underlying mechanisms involved. Preconditioning with 40-160 mg/kg, (S)-ZJM-289 significantly reduces brain damage after I/R. (S)-ZJM-289 preconditioning is effective when applied 1-3 days before I/R. Moreover, the degrees of neuroprotection offered by (S)-ZJM-289 preconditioning and ischemic preconditioning are virtually identical. (S)-ZJM-289 preconditioning also protects primary cultured cortical neurons against oxygen-glucose deprivation and recovery-induced cytotoxicity in vitro. (S)-ZJM-289 preconditioning significantly increases the generation of NO, but has no effect on the nitric oxide synthase activities. Additionally, (S)-ZJM-289 preconditioning promotes the dissociation between nuclear-factor-E2-related factor (Nrf2) and kelch-like ECH-associated protein 1, and induces Nrf2 nuclear localization. The neuroprotection of (S)-ZJM-289 preconditioning is blocked by Nrf2-siRNA in vitro. (S)-ZJM-289 preconditioning up-regulates antioxidant enzymes against nervous injury. (S)-ZJM-289 preconditioning significantly activates extracellular regulated protein kinases (ERK) and inhibits c-Jun N-terminal kinases signaling cascade. The neuroprotection is abolished by the ERK inhibitor PD98059 in vitro. Subsequently, (S)-ZJM-289 preconditioning increases the levels of anti-apoptotic protein B cell lymphoma 2 (Bcl-2) and inhibited the translocation of Bcl-2 associated X to the mitochondria, thus attenuating the release of cytochrome c from the mitochondria and the activation of downstream caspase. These results suggest that (S)-ZJM-289 preconditioning exerts the late phase protection against nervous injury induced by transient cerebral ischemia and oxygen-glucose deprivation.
SALL4, a zinc-finger transcriptional factor for embryonic stem cell self-renewal and pluripotency, has been suggested to be involved in tumorigenesis. The role of SALL4 in human gastric cancer, however, remains largely unknown. In this study, we demonstrated that SALL4 was aberrantly expressed at both mRNA and protein levels in human gastric cancer tissues, and SALL4 level was highly correlated with lymph node metastasis. Enforced expression of SALL4 enhanced the proliferation and migration of human gastric cancer cells, whereas knockdown of SALL4 by siRNA led to the opposite effects. In addition, SALL4 overexpression promoted the growth and metastasis of gastric xenograft tumor in vivo. SALL4 overexpression induced epithelial-mesenchymal transition (EMT) in gastric cancer cells, with increased expression of Twist1, N-cadherin and decreased expression of E-cadherin. Moreover, SALL4 promoted the acquirement of stemness in gastric cancer cells through the induction of Bmi-1 and Lin28B. Taken together, our findings indicate that SALL4 has oncogenic roles in gastric cancer through the modulation of EMT and cell stemness, suggesting SALL4 as a novel target for human gastric cancer diagnosis and therapy.
2,2',4,4'-tetrabromodiphenyl ether (PBDE-47)-elicited neurotoxicity is associated with neural apoptosis; however the underlying mechanisms remain unclear. To investigate whether the mitochondrial p53 pathway is involved in neuronal apoptosis induced by PBDE-47 and to correlate DNA hypomethylation with p53 activation, human neuroblastoma (SH-SY5Y) cells were treated with different concentrations of PBDE-47 (1, 5, 10 μmol/L) for 24h in vitro. The apoptosis and ultrastructural alterations in cells, levels of p53, Bcl-2, Bax, cytochrome c (Cyt c), caspase-3 and methylation status of p53 promoter were determined. Hoechst 33258 staining and transmission electron microscopy analysis showed that PBDE-47 induced SH-SY5Y cells apoptosis characterized by the typical apoptotic morphological changes. In addition, PBDE-47 activated the p53-dependent mitochondrial apoptotic pathway as evidenced by up-regulation of p53 and Bax, down-regulation of Bcl-2 and Bcl-2/Bax ration, enhancement of Cyt c release from mitochondria into the cytosol, activation of caspase-3 as well as ultrastructural abnormalities of mitochondria. However, no obvious decrease in p53 promoter methylation levels was observed in any of the treatment groups by bisulfite genomic sequencing. Collectively, these results suggest that the mitochondrial p53 pathway is involved in PBDE-47-induced SH-SY5Y cells apoptosis, nevertheless p53 promoter hypomethylation may not be implicated in this process.
Reactive oxygen species (ROS)-induced oxidative stress increases in skeletal muscle with aging and decreases the viability of implanted cells. Type 1 insulin-like growth factor (IGF-1) promotes the survival of skeletal muscle cells under oxidative stress. It is unknown whether IGF-1 protects muscle-derived stem cells (MDSCs) from oxidative stress. In this study, we genetically engineered rat MDSCs to overexpress IGF-1 and determined cell viability, apoptosis, and VEGF secretion under oxidative stress. Overexpression of IGF-1 prevented MDSCs from H2O2-induced caspase-dependent apoptotic cell death by upregulating the PI3K/AKT pathway, accompanied with an increase of NF-κB, p-NF-κB, Bcl-2, and VEGF, as well as a decrease of Bax. In contrast, pre-administration of picropodophyllinb, wortmannin, 1L-6-hydroxymethyl-chiro-inositol-2-((R)-2-O-methyl-3-O-octadecylcarbonate), or pyrrolidine-dithiocarbamate, specific inhibitors of IGF-1R, PI3K, AKT, and NF-κB, respectively, followed by treatment with H2O2, resulted in cell death of MDSCs. Our data indicated that IGF-1 suppresses apoptosis and enhances the paracrine function of MDSCs under oxidative stress via enhancing IGF-1R/PI3K/AKT signaling. Thus, IGF-1 gene-modified MDSCs present a potential application in the treatment of muscle wasting, such as urethra intrinsic sphincter deficiency.
Necroptosis was recently discovered as one form of programmed cell death (PCD) and could be specifically inhibited by necrostatin-1. The aim of this study was to examine the effect of necrostatin-1 on brain injury and investigate the role of necrostatin-1 on the other two types PCD (apoptosis and autophagic cell death) in a mouse intracerebral hemorrhage (ICH) model. Male ICR mice received an infusion of type IV collagenase to induce ICH or saline as control into the left striatum. In the presence of vehicle, 3-MA, zVAD, and necrostatin-1 were pretreated with a single intracerebroventricular (i.c.v.) injection in the ipsilateral ventricle 15 min before ICH, respectively. Compared with vehicle groups, necrostatin-1 treatment significantly reduced injury volume and propidium iodide-positive cells at 24 and 72 h after ICH. Immunoblotting analysis showed that necrostatin-1 treatment suppressed autophagic-associated proteins (LC3-II, Beclin-1) and maintained p62 at normal level at 24 and 72 h after ICH. In addition, necrostatin-1 treatment enhanced the protein level of Bcl-2 and decreased the protein level of cleaved caspase-3 and the Beclin-1/Bcl-2 ratio at 24 and 72 h after ICH. Moreover, both 3-MA and necrostatin-1 treatment could suppress cleaved caspase-3 and LC3-II production, whereas zVAD treatment could inhibit caspase-3 cleavage but increased LC3-II protein levels at 72 h after ICH. Taken together, the data demonstrated for the first time that the specific inhibitor necrostatin-1 suppressed apoptosis and autophagy to exert these neuroprotective effects after ICH and that there existed a cross-talk among necroptosis, apoptosis, and autophagy after ICH.
Mesenchymal stem cells (MSCs) have been considered as an attractive tool for the therapy of diseases. Exosomes excreted from MSCs can reduce myocardial ischemia/reperfusion damage and protect against acute tubular injury. However, whether MSC-derived exosomes can relieve liver fibrosis and its mechanism remain unknown. Previous work showed that human umbilical cord-MSCs (hucMSCs) transplanted into acutely injured and fibrotic livers could restore liver function and improve liver fibrosis. In this study, it was found that transplantation of exosomes derived from hucMSC (hucMSC-Ex) reduced the surface fibrous capsules and got their textures soft, alleviated hepatic inflammation and collagen deposition in carbon tetrachloride (CCl4)-induced fibrotic liver. hucMSC-Ex also significantly recovered serum aspartate aminotransferase (AST) activity, decreased collagen type I and III, transforming growth factor (TGF)-β1 and phosphorylation Smad2 expression in vivo. In further experiments, we found that epithelial-to-mesenchymal transition (EMT)-associated markers E-cadherin-positive cells increased and N-cadherin- and vimentin-positive cells decreased after hucMSC-Ex transplantation. Furthermore, the human liver cell line HL7702 underwent typical EMT after induction with recombinant human TGF-β1, and then hucMSC-Ex treatment reversed spindle-shaped and EMT-associated markers expression in vitro. Taken together, these results suggest that hucMSC-Ex could ameliorate CCl4-induced liver fibrosis by inhibiting EMT and protecting hepatocytes. This provides a novel approach for the treatment of fibrotic liver disease.
The lethal effects of freezing on cells are currently attributed to the crystallization of extracellular water which leaves concentrated solutions of salts, macromolecules and so forth in the extracellular space. This concentrated fluid establishes a strong osmotic gradient which draws water from the cells. Thus, a cell surrounded by ice can survive only if means can be found for reducing the osmotically driven outflow of cellular water. This is usually attempted through vitrification of the extracellular space, but may also be attained through suitable modifications of cellular plasms. Starting from microscopic observations on early rabbit embryos and related cryotolerance, we investigated purified actin solutions under similar conditions, and found that sol-gel processing could result in the formation of homogeneous glass, and through drying, give rise to monolithic solids, glasses and composites. The first process may be at least partially responsible for the induced cryotolerance of cells, while the second may be representative of new and useful biomaterials.
SCOPE:
In this study, we focus on the effects of n-3 polyunsaturated fatty acids (PUFAs) on tunicamycin-, streptozotocin-, or high fat diet (HFD)-induced β-cell damage and dysfunction.
MATERIALS AND METHODS:
Pretreatment with n-3 PUFAs protected RINm5F cells and mouse islets against tunicamycin-induced β-cell damage through suppression of ER stress and apoptosis induction. This protective effect of n-3 PUFAs on β-cells was further demonstrated by the normalization of insulin secretion in response to glucose in tunicamycin-treated islets. In multiple low-dose streptozotocin-induced diabetes models, fat-1 mice, which endogenously synthesize n-3 PUFAs from n-6 PUFAs, were fully resistant to the development of diabetes, with normal islet morphology, high insulin immunoreactivity, and decreased apoptotic cells. In HFD-induced diabetes models, fat-1 mice also exhibited improved glucose tolerance and functional β-cell mass. In both diabetes models, we observed an attenuation of ER stress in fat-1 mice. Interestingly, n-3 PUFAs attenuated the nuclear translocation of lipogenic transcription factors sterol regulatory element-binding protein-1 (SREBP-1) and C/EBPβ, induced by tunicamycin or HFD, suggesting that n-3 PUFAs suppress ER stress via modulation of SREBP-1 and C/EBPβ.
CONCLUSION:
Together, these results suggest that n-3 PUFAs block ER stress, thus protecting β cells against diabetogenic insult; therefore, dietary supplementation of n-3 PUFAs has therapeutic potential for the preservation of functional β-cell mass.
© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
BACKGROUND:
Paclitaxel (PTX) is a first line chemotherapy drug for breast cancer. There have been few studies reported concerning the therapeutic efficacy of paclitaxel-conjugated polymeric micelles in breast cancer in vivo.
METHODS:
Two kinds of PTX conjugate micelles, one of which (M(PTX)) contained 25 wt.% of PTX and the other (M(FA/PTX)) contained 22.5 wt.% of PTX and 1.4 wt.% of folate (FA), were prepared for cell apoptosis and anti-tumor activity evaluation on EMT-6 mice breast cancer models in comparison with 0.9 wt.% saline (control) and equivalent PTX. Cell apoptosis was analyzed by flow cytometry. Breast tumors were examined histologically with H&E staining and immunohistochemically by examining Bax and Bcl-2 expression. The survival status of tumor-bearing mice with different treatments was also examined.
RESULTS:
On day 5 of the drug administration, the average tumor masses were 0.49, 0.33, 0.22, and 0.18 g for the control, PTX, M(PTX) and M(FA/PTX) groups, respectively. The inhibition rates of tumor growth calculated for the three drug groups were 32.6%, 51.6% and 62.3%, respectively. The percentage of cell apoptosis based on flow cytometry was 1.0%, 36.6%, 55.9% and 66.1%, respectively, which showed statistically significant differences (p<0.05) between three drug groups and the control group. Bcl-2 expression of PTX and M(FA/PTX) groups was lower than control group (p<0.05). Bax expression of drug groups was higher than control group (p<0.05). At an equivalent paclitaxel dose of 26.7 mg/kg, the average survival time was 33 days, 31 days, 34 days and 42 days, respectively (p<0.05).
CONCLUSION:
The M(FA/PTX) have better anti-tumor activity and are promising in treatment of human breast cancers.
Copyright © 2015 Elsevier B.V. All rights reserved.
OBJECTIVE:
Local electrical stimulation (ES) was reported to protect the brain during ischaemic injury, while the protective effect of limb remote ischaemic postconditioning (RIPostC) was confirmed. The aim of this study was to explore whether remote peripheral nerve ES exerted neuroprotection and whether this procedure shared the same neuroprotective mechanism underlying RIPostC.
METHODS:
Stroke in Sprague-Dawley rats was induced by distal middle cerebral artery occlusion (dMCAO). Rats were divided into five groups: dMCAO, RIPostC, ES, nerve resection (NR) + ES and RIPostC+ES. Twenty-four hours after reperfusion, rats were examined for neurobehavioural function, including forelimb fault placing test, Ludmila Belayev 12 score test, and infarct volume. The expression of Bcl-2 and cleaved-caspase-3 in ischaemic cortex was assessed by Western blot.
RESULTS:
In forelimb fault placing test, as compared to the highest score in the stroke-only group, RIPostC, ES and RIPostC+ES groups showed a significantly (P < 0.01) lower score. The results were similar for the Ludmila Belayev 12 score test. The infarct volume of the treatment groups also exhibited significant (P < 0.01) reduction as compared to the stroke-only group. The volume of infarct tissue in the combination of RIPostC+ES was significantly less than RIPostC and ES alone (P < 0.05). Furthermore, NR blocked the ES's protection (P < 0.05) as compared to the ES group by using above-mentioned methods. Bcl-2 was upregulated, while cleaved-caspase-3 was downregulated in the experimental groups as compared to the control group. No difference was found among the experimental groups.
DISCUSSION:
Peripheral nerve ES appears to have a neuroprotective effect in a rat dMCAO model. This effect may indicate a neural protective mechanism underlying beneficial effect of RIPostC.
AIMS:
This study aimed to investigate the pathogenesis mechanisms of bleomycin (BLM)-induced pulmonary fibrosis (PF) in Sprague-Dawley rats and explore the anti-fibrotic role of arsenic trioxide (As2O3) in preventing PF.
MAIN METHODS:
Intratracheal instillation of BLM was performed to establish PF rat models. The treatment group was treated with As2O3 (0.4 mg/kg/day). Morphological changes were observed by hematoxylin-eosin and Masson staining. Related proteins were determined by immunohistochemistry, immunofluorescence, and Western blot. MicroRNA detection was performed by quantitative real-time polymerase chain reaction.
KEY FINDINGS:
As a novel miRNA in PF, miR-98 decreased in the fibrotic lung tissues. Based on microRNA analysis software, we found that Stat3-3'-UTR is targeted by miR-98. Then, we found that Stat3 was activated with PF development and the expression of Stat3 and p-Stat3 was significantly increased in BLM-induced PF at day 28 compared with saline-treated rats. Our results showed that both Stat3 and p-Stat3 were significantly decreased in miR-98-treated A549 cells compared with that in mu-98-treated cultures or untreated controls. The fibrotic marker α-SMA was upregulated, whereas E-cadherin was inhibited in fibrotic lung tissues. The ratio of apoptotic factors Bax/Bcl-2 increased with the development of fibrosis. Furthermore, As2O3 treatment prevented lung interstitial thickening and inhibited the collagen type I and hydroxyproline, thereby preventing the development of PF. As2O3 also significantly down-regulated α-SMA but increased E-cadherin and miR-98 levels.
SIGNIFICANCE:
The study revealed that arsenic trioxide prevented rat PF by up-regulation of miR-98 and inhibition of its downstream Stat3 signals.
Copyright © 2014 Elsevier Inc. All rights reserved.
BACKGROUND INFORMATION:
Microtubule affinity-regulating kinase 4 (MARK4) deficiency has been reported to negatively regulate diet-induced obesity and to mitigate insulin resistance in knockout mice, and thus may play a role in metabolic syndrome. However, the details of the molecular mechanism have yet to be revealed and the impacts of MARK4 on apoptosis remain unexplored. This study investigated the role of Mark4 in the regulation of lipid accumulation and apoptosis in adipocytes and analysed signalling pathways involved.
RESULTS:
We found that Mark4 significantly up-regulated the expression of gene sterol regulatory element binding protein-1c (SREBP-1c), fatty acid synthase (FAS), acetyl-CoA carboxylase-α (ACCα) and peroxisome proliferator activated receptor-γ (PPARγ); and reduced the protein contents of adipose triglyceride lipase (ATGL), as evidenced by the dramatic increasing lipid droplet accumulation in 3T3-L1 cells. Furthermore, a terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) apoptosis assay showed that Mark4 triggered apoptosis of adipocytes; and apoptosis was confirmed by the decreased protein contents of B-cell lymphoma-2 (Bcl-2), full-length caspase-3 and full-length caspase-9, as well as the increased expression of Bax, cleaved caspase-3 and cleaved caspase-9. Analysis of special inhibitors allowed us to offer the following explanation for these impacts of Mark4: activation of Jun N-terminal kinase1 (JNK1) promoted both apoptosis and adipogenesis, whereas inhibition of the p38 mitogen-activated protein kinase (p38MAPK) pathway contributed to lipid accumulation alone.
CONCLUSIONS:
Mark4 promotes adipogenesis in 3T3-L1 adipocytes by activating the JNK1 and inhibiting the p38MAPK pathway, and triggers apoptosis by activating the JNK1 pathway. We conclude that anti-Mark4 therapy targetted to inhibit lipid accumulation and apoptosis of adipocytes shows potential as a novel therapeutic strategy for treatment of obesity-associated metabolic complications.
© 2014 Société Française des Microscopies and Société de Biologie Cellulaire de France. Published by John Wiley & Sons Ltd.
PURPOSE:
This study was designed to investigate the role of PDGF-DD secreted by gastric cancer-derived mesenchymal stem cells (GC-MSCs) in human gastric cancer progression.
METHODS:
Gastric cancer cells were indirectly co-cultured with GC-MSCs in a transwell system. The growth and migration of gastric cancer cells were evaluated by cell colony formation assay and transwell migration assay, respectively. The production of PDGF-DD in GC-MSCs was determined by using Luminex and ELISA. Neutralization of PDGFR-β by su16f and siRNA interference of PDGF-DD in GC-MSCs was used to demonstrate the role of PDGF-DD produced by GC-MSCs in gastric cancer progression.
RESULTS:
GC-MSC conditioned medium promoted gastric cancer cell proliferation and migration in vitro and in vivo. Co-culture with GC-MSCs increased the phosphorylation of PDGFR-β in SGC-7901 cells. Neutralization of PDGFR-β by su16f blocked the promoting role of GC-MSC conditioned medium in gastric cancer cell proliferation and migration. Recombinant PDGF-DD duplicated the effects of GC-MSC conditioned medium on gastric cancer cells. Knockdown of PDGF-DD in GC-MSCs abolished its effects on gastric cancer cells in vitro and in vivo.
CONCLUSIONS:
PDGF-DD secreted by GC-MSCs is capable of promoting gastric cancer cell progression in vitro and in vivo. Targeting the PDGF-DD/PDGFR-β interaction between MSCs and gastric cancer cells may represent a novel strategy for gastric cancer therapy.
ETHNOPHARMACOLOGICAL RELEVANCE:
Nephrotic syndrome (NS) is a clinical syndrome with a variety of causes, mainly characterized by heavy proteinuria. Podocyte injury plays a key role in proteinuria, one of the principal means for the control of NS is to prevent podocyte injury. Qi-Dan Fang consists of two of the most extensively applied herbal remedies among Traditional Chinese Medicine (TCM) (Radix Astragali Mongolici and Radix Salviae Miltiorrhizae, with a weight ratio of 5:1) which are specifically used for the treatment of various kidney diseases. In previous studies, we found that Qi-Dan Fang provides improvement to patients with adriamycin-induced nephrotic syndrome by alleviating proteinuria and serum lipid. The aim of this study is to study the efficiency of Qi-Dan Fang on NS model rat with renal dysfunction and podocyte injury, something which has not been carried out yet.
MATERIALS AND METHODS:
The rats were divided into Normal, Model, Jin Gui Shen Qi Pill (4.12 g/kg), Qi-Dan Fang (3.09, 6.17 and 12.34 g/kg/d) groups, they were each given a single tail intravenous injection of Adriamycin (6.0 mg/kg) except for the Normal group and were orally administered dosages of Qi-Dian Fang and Jin Gui Shen Qi pills once daily for 7 weeks. Following the treatment, the content of cystation C (CysC), blood urea nitrogen (BUN), serum creatinine (Scr) were measured with an autobiochemical analyser. The pathomorphological changes to the glomeruli, the mRNA expressions of nephrin, podocin, CD2AP genes and p53, bax, bcl-2 proteins expressions were also carried out to probe the effects of Qi-Dan Fang.
RESULTS:
(1) Qi-Dan Fang treatment raised the level of CysC in blood serum while lowering the content of BUN and Scr in the adriamycin-induced nephrotic syndrome rat model; (2) Long-term administration of Qi-Dan Fang was able to ameliorate pathomorphological change of glomeruli and repair the organization structure of Glomerulus; (3) Qi-Dan Fang could increase the mRNA expression of nephrin, podocin and CD2AP genes, down-regulate the expression of p53, bax proteins, while increased bcl-2 protein to protect the podocyte and restore Glomerular selective filtration function.
CONCLUSIONS:
Results of our present studies reveal that Qi-Dan Fang is able to enhance renal function, inhibit podocyte injury to provide improvements to the Adriamycin-induced nephrotic syndrome.
Copyright © 2013 Elsevier Ireland Ltd. All rights reserved.

![Western Blot - Anti-Bcl-2 Antibody [AMC0441] (A309289) - Antibodies.com](https://cdn.antibodies.com/image/catalog/309/A309289_1.jpg?profile=product_alternative)
![Western Blot - Anti-Bcl-2 Antibody [ARC0173] (A306910) - Antibodies.com](https://cdn.antibodies.com/image/catalog/306/A306910_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Bcl-2 Antibody [100/D5+124] (A249868) - Antibodies.com](https://cdn.antibodies.com/image/catalog/249/A249869_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Bcl-2 Antibody [100/D5] - BSA and Azide free (A253043) - Antibodies.com](https://cdn.antibodies.com/image/catalog/253/A253044_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Bcl-2 Antibody [100/D5] (A249863) - Antibodies.com](https://cdn.antibodies.com/image/catalog/249/A249864_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Bcl-2 Antibody [rBCL2/796] - BSA and Azide free (A253053) - Antibodies.com](https://cdn.antibodies.com/image/catalog/253/A253053_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Bcl-2 Antibody [rBCL2/796] (A249873) - Antibodies.com](https://cdn.antibodies.com/image/catalog/249/A249873_1.jpg?profile=product_alternative)