Primary Antibodies
Secondary Antibodies
Proteins & Peptides
ELISA Kits
About Us
Contact Us
Sign In/Register
0
ISO 9001:2015 Certified
Live Customer Support
4.5/5 on Trustpilot
100% Quality Guarantee
Home
Primary Antibodies
Calnexin Antibodies
Anti-Calnexin Antibody (A121570)
Anti-Calnexin Antibody (A121570)
Overview
Specifications
Images
Enlarge Image
Enlarge Image
$590
Product Datasheet
Goat polyclonal antibody to Calnexin for WB, IF, IHC-P and IHC-Fr.
100% Guarantee
Price Match Guarantee
Product Size:
400µg
1mg
Quantity:
1
2
3
4
5
6
7
8
9
10
Add To Cart
Request a Quotation
Custom or Bulk Request
Shipping Information
Freight/Packing Charges:
$40
Dispatched from St. Louis, MO.
Lead Time: 5-8 business days.
Tags:
ER Membrane Marker
Ribosomal Marker
Specifications
Name
Anti-Calnexin Antibody
Description
Goat polyclonal antibody to Calnexin.
Applications
WB
,
IF
,
IHC-P
,
IHC-Fr
Dilutions
WB: 1:500-1:5,000, IF: 1:50-1:500, IHC-P: 1:200-1:1,000, IHC-Fr: 1:200-1:1,000
Reactivity
Human, Rat, Mouse, Canine, Monkey
Immunogen
Recombinant peptide derived from within residues a.a. 550 to the C terminus of human CANX, expressed in and purified from E. coli.
Host
Goat
Clonality
Polyclonal
Isotype
IgG
Conjugate
Unconjugated
Purification
Affinity purification.
Concentration
2 mg/ml
Product Form
Liquid
Formulation
Supplied in Phosphate Buffered Saline with 20% Glycerol and 0.05% Sodium Azide.
Storage
Shipped at 4°C. Upon delivery aliquot and store at -20°C. Avoid freeze/thaw cycles.
Synonyms
CANX, IP90, Major histocompatibility complex class I antigen-binding protein p88, p90
Isotype Controls
Goat IgG (A121671)
Suitable Secondaries
Donkey Anti-Goat IgG H&L Antibody (AP) (A300679)
Donkey Anti-Goat IgG H&L Antibody (Biotin) (A300716)
Donkey Anti-Goat IgG H&L Antibody (FITC) (A300685)
Donkey Anti-Goat IgG H&L Antibody (HRP) (A300730)
See all Anti-Goat IgG Secondaries →
Disclaimer
This product is for research use only. It is not intended for diagnostic or therapeutic use.
Scientific Validation Data
Validation Data
(2)
Enlarge Image
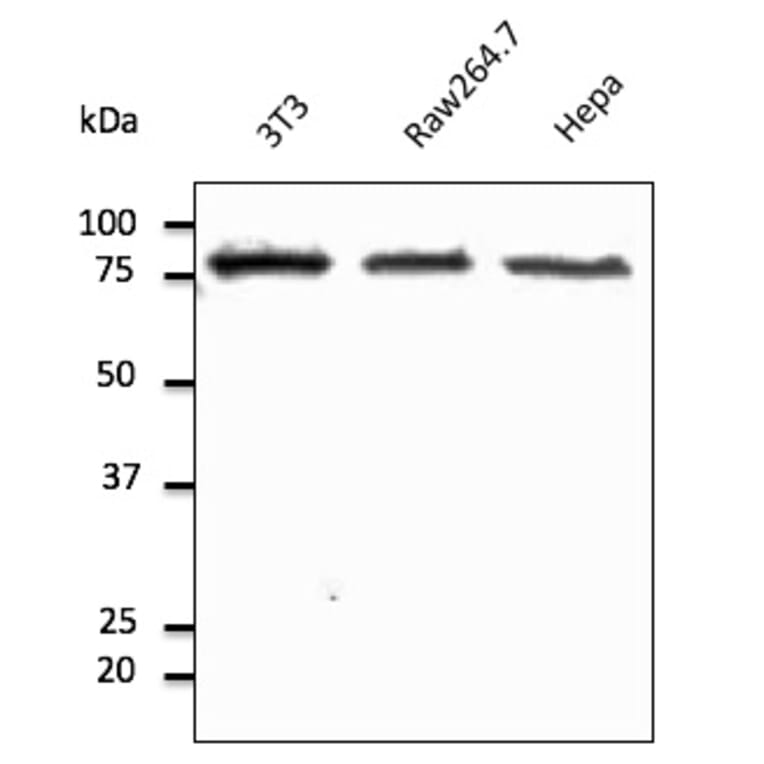
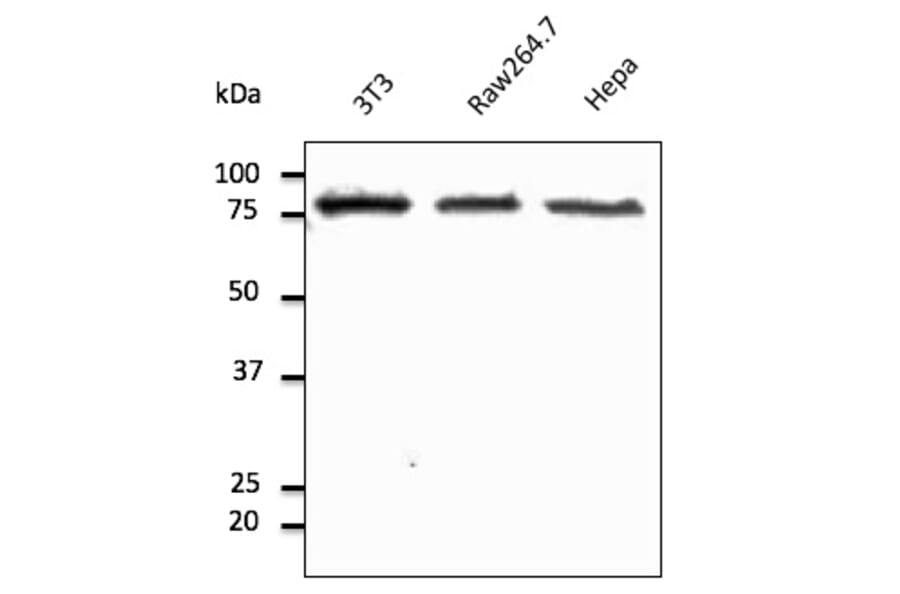
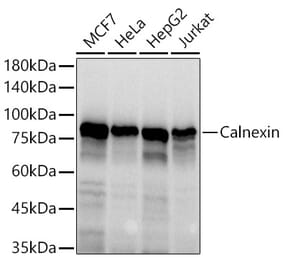
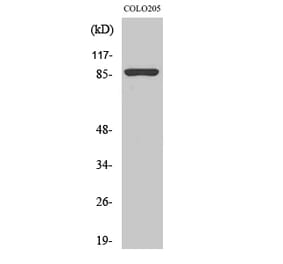
Western Blot - Anti-Calnexin Antibody (A121570)
HeLa cell lysate detected with Anti-Calnexin Antibody at a 1:2,500 dilution. Lysate at 50µg and rabbit anti-goat IgG antibody (HRP) at a 1:10,000 dilution.
Enlarge Image
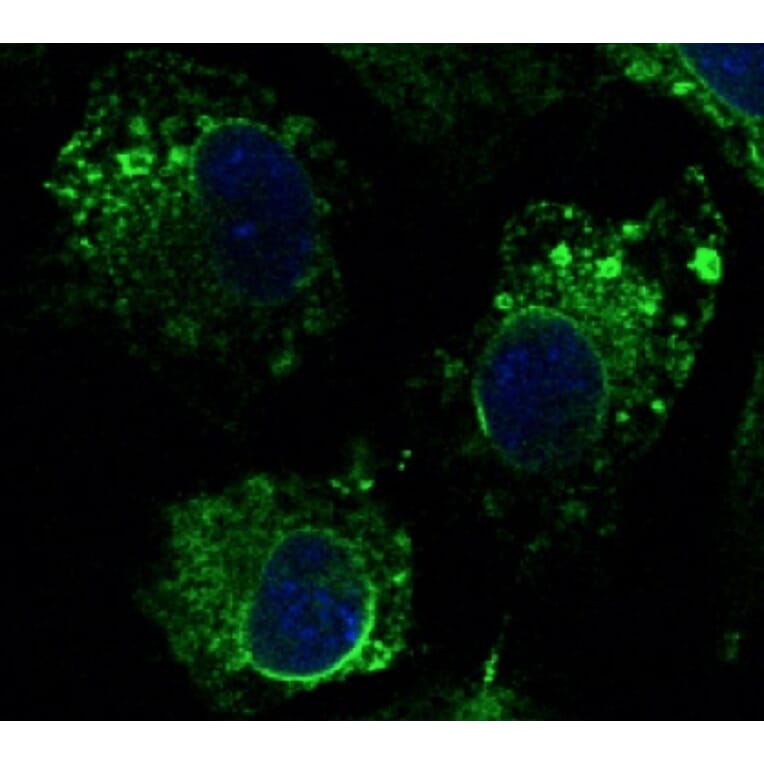
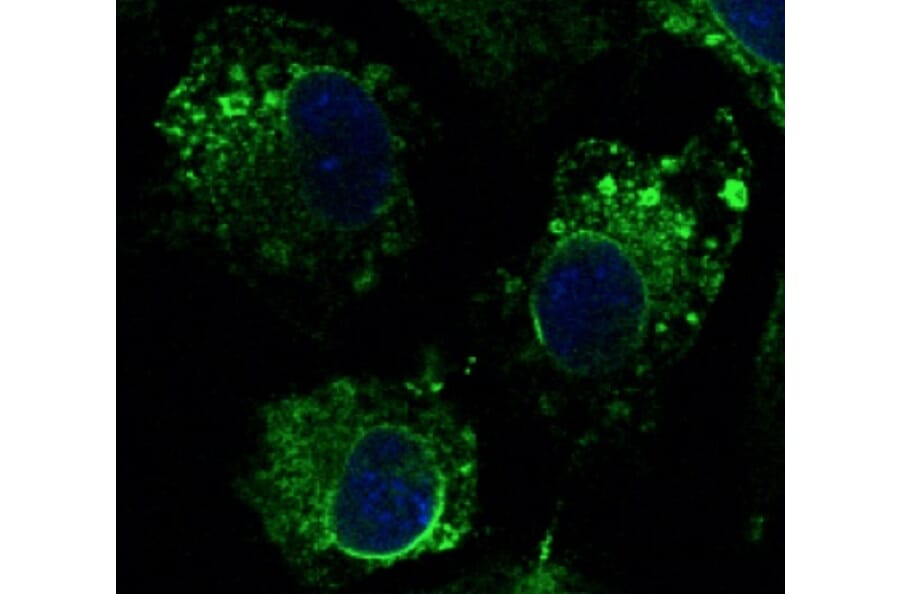
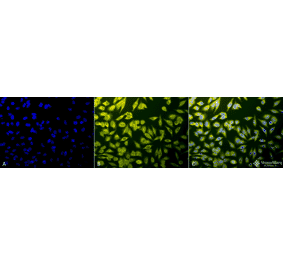
Anti-Calnexin Antibody (A121570)
Hepa 1-6 cells, fixed with 4% PFA, stained with Anti-Calnexin Antibody at a 1:100 dilution.
Publishing research using Anti-Calnexin Antibody (A121570)? Please
let us know
so that we can list the citation on this page.
Most popular Anti-Calnexin Antibodies
(9)
A94879
Anti-Calnexin Antibody
Rabbit polyclonal antibody to Calnexin for WB, IHC, IF and ELISA.
(6)
A83665
Anti-Calnexin Antibody
Goat polyclonal antibody to Calnexin for ELISA, WB, IHC and IF.
(6)
A429
Anti-Calnexin Antibody
Rabbit polyclonal antibody to Calnexin for WB, IP, IF and IHC.
(9)
A94879
Anti-Calnexin Antibody
Rabbit polyclonal antibody to Calnexin for WB, IHC, IF and ELISA.
(6)
A83665
Anti-Calnexin Antibody
Goat polyclonal antibody to Calnexin for ELISA, WB, IHC and IF.
(6)
A429
Anti-Calnexin Antibody
Rabbit polyclonal antibody to Calnexin for WB, IP, IF and IHC.
Alternative products to Anti-Calnexin Antibody (A121570)
(5)
A305107
Anti-Calnexin Antibody
Rabbit polyclonal antibody to Calnexin for WB, IHC, ICC/IF, IP and Flow Cytometry.
(4)
A305106
Anti-Calnexin Antibody
Rabbit polyclonal antibody to Calnexin for WB, ICC/IF and IHC.
A304551
Anti-Calnexin Antibody
Rabbit polyclonal antibody to Calnexin for WB.
(4)
A87725
Anti-Calnexin Antibody
Rabbit polyclonal antibody to Calnexin for WB, ICC/IF and IP.
(2)
A25463
Anti-Calnexin (D577) Antibody
Rabbit polyclonal antibody to Calnexin (D577) for WB, IHC and IF.
(4)
A94794
Anti-Calnexin (phospho Ser583) Antibody
Rabbit polyclonal antibody to Calnexin (phospho Ser583) for WB, IHC, IF and ELISA.
(2)
A121572
Anti-Calnexin Antibody
Goat polyclonal antibody to Calnexin for WB, IF, IHC-P and IHC-Fr.
(7)
A250400
Anti-Calnexin Antibody [CANX/1541]
Mouse monoclonal [CANX/1541] antibody to Calnexin for ELISA, WB and IHC-P.
(7)
A253580
Anti-Calnexin Antibody [CANX/1541] - BSA and Azide free
Mouse monoclonal [CANX/1541] antibody to Calnexin for ELISA, WB and IHC-P.
(5)
A250401
Anti-Calnexin Antibody [CANX/1543]
Mouse monoclonal [CANX/1543] antibody to Calnexin for ELISA, WB and IHC-P.
(2)
A34321
Anti-Calnexin Antibody
Rabbit polyclonal antibody to Calnexin for WB, IHC, ICC/IF and ELISA.
(5)
A253581
Anti-Calnexin Antibody [CANX/1543] - BSA and Azide free
Mouse monoclonal [CANX/1543] antibody to Calnexin for ELISA, WB and IHC-P.
(5)
A305107
Anti-Calnexin Antibody
Rabbit polyclonal antibody to Calnexin for WB, IHC, ICC/IF, IP and Flow Cytometry.
(4)
A305106
Anti-Calnexin Antibody
Rabbit polyclonal antibody to Calnexin for WB, ICC/IF and IHC.
A304551
Anti-Calnexin Antibody
Rabbit polyclonal antibody to Calnexin for WB.
(4)
A87725
Anti-Calnexin Antibody
Rabbit polyclonal antibody to Calnexin for WB, ICC/IF and IP.
(2)
A25463
Anti-Calnexin (D577) Antibody
Rabbit polyclonal antibody to Calnexin (D577) for WB, IHC and IF.
(4)
A94794
Anti-Calnexin (phospho Ser583) Antibody
Rabbit polyclonal antibody to Calnexin (phospho Ser583) for WB, IHC, IF and ELISA.
(2)
A121572
Anti-Calnexin Antibody
Goat polyclonal antibody to Calnexin for WB, IF, IHC-P and IHC-Fr.
(7)
A250400
Anti-Calnexin Antibody [CANX/1541]
Mouse monoclonal [CANX/1541] antibody to Calnexin for ELISA, WB and IHC-P.
(7)
A253580
Anti-Calnexin Antibody [CANX/1541] - BSA and Azide free
Mouse monoclonal [CANX/1541] antibody to Calnexin for ELISA, WB and IHC-P.
(5)
A250401
Anti-Calnexin Antibody [CANX/1543]
Mouse monoclonal [CANX/1543] antibody to Calnexin for ELISA, WB and IHC-P.
(2)
A34321
Anti-Calnexin Antibody
Rabbit polyclonal antibody to Calnexin for WB, IHC, ICC/IF and ELISA.
(5)
A253581
Anti-Calnexin Antibody [CANX/1543] - BSA and Azide free
Mouse monoclonal [CANX/1543] antibody to Calnexin for ELISA, WB and IHC-P.
See all Calnexin Antibodies
Proteins predicted to interact with Calnexin
Predicted protein interactions based upon String database. Revelancy score correlates with probability of interaction.
ERp57 Antibodies
99.9% Relevancy Score
Calreticulin Antibodies
99.9% Relevancy Score
BIP Antibodies
99.8% Relevancy Score
Tapasin Antibodies
99.6% Relevancy Score
GRP94 Antibodies
99% Relevancy Score
EDEM1 Antibodies
98.6% Relevancy Score
VCP Antibodies
96.7% Relevancy Score
P4HB Antibodies
96.6% Relevancy Score
OS9 Antibodies
95.8% Relevancy Score
Top





![Immunohistochemistry - Anti-Calnexin Antibody [CANX/1541] (A250400) - Antibodies.com](https://cdn.antibodies.com/image/catalog/250/A250400_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Calnexin Antibody [CANX/1541] - BSA and Azide free (A253580) - Antibodies.com](https://cdn.antibodies.com/image/catalog/253/A253580_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Calnexin Antibody [CANX/1543] (A253580) - Antibodies.com](https://cdn.antibodies.com/image/catalog/250/A250401_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Calnexin Antibody [CANX/1543] - BSA and Azide free (A250401) - Antibodies.com](https://cdn.antibodies.com/image/catalog/253/A253581_1.jpg?profile=product_alternative)