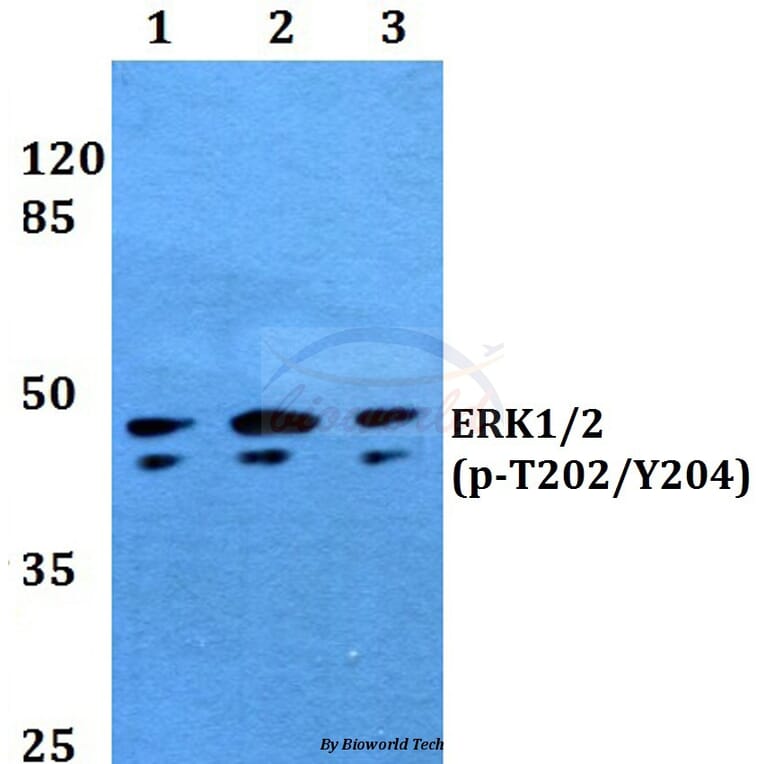
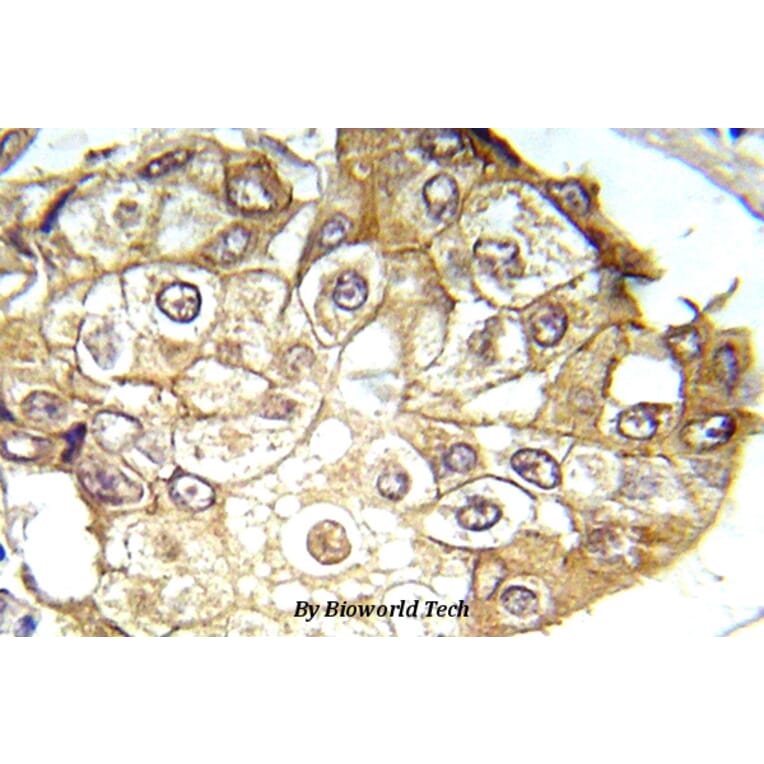
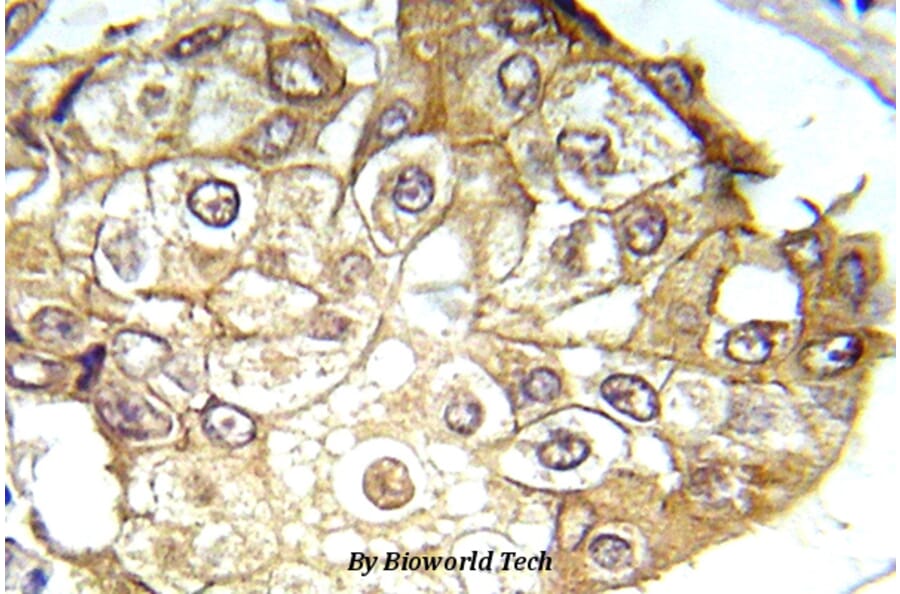
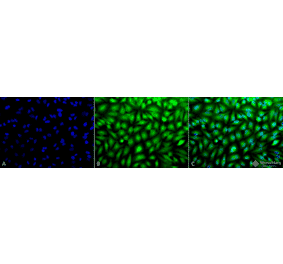
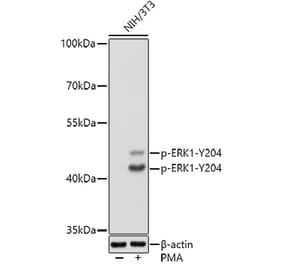
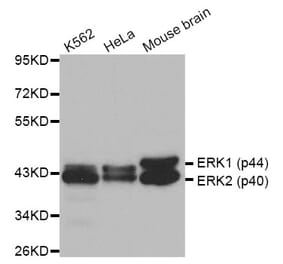
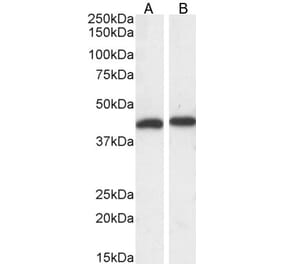
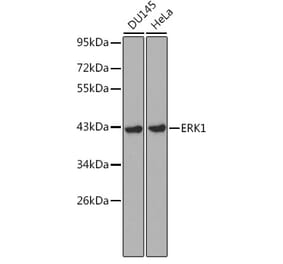
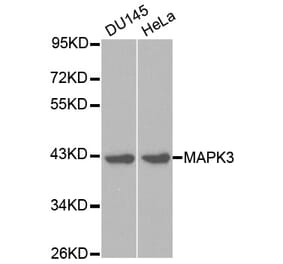
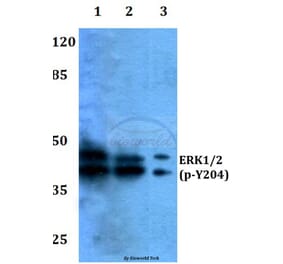
Synonyms
ERK-1, ERT2, Extracellular signal-regulated kinase 1, Insulin-stimulated MAP2 kinase, MAP kinase 3, MAP kinase isoform p44, MAPK 3, MAPK3, Microtubule-associated protein 2 kinase, Mitogen-activated protein kinase 3, p44-ERK1, p44-MAPK, PRKM3