Laura Norton, PhD | 15th April 2020
Insulin is a peptide hormone that predominantly functions to reduce blood glucose levels. It is secreted from beta cells found in the islets of the pancreas in response to nutrient uptake and increased blood glucose levels. When insulin binds to its receptors on target cells, such as skeletal muscle cells and adipocytes, a signaling cascade is initiated, which culminates in the translocation of the glucose transporter GLUT4 from intracellular vesicles to the cell membrane. Once GLUT4 is incorporated into the plasma membrane, it functions to promote the uptake of extracellular glucose, which is then stored as glycogen in these cells, thereby regulating blood glucose [1].
Insulin also regulates blood sugar through inhibiting gluconeogenesis (de novo glucose production) and glycogenolysis (glycogen breakdown) in the liver. Besides regulating blood glucose levels, insulin also plays critical roles in facilitating protein and lipid synthesis and preventing the conversion of protein and fat to glucose.
While insulin is widely viewed as a glucose homeostasis regulating hormone, an increasing body of research is illuminating broader roles for this peptide. Insulin signaling pathways are highly conserved, with insulin-like signaling systems found in all metazoans, and they have been shown to regulate many evolutionarily conserved processes, including lifespan and reproduction [2].
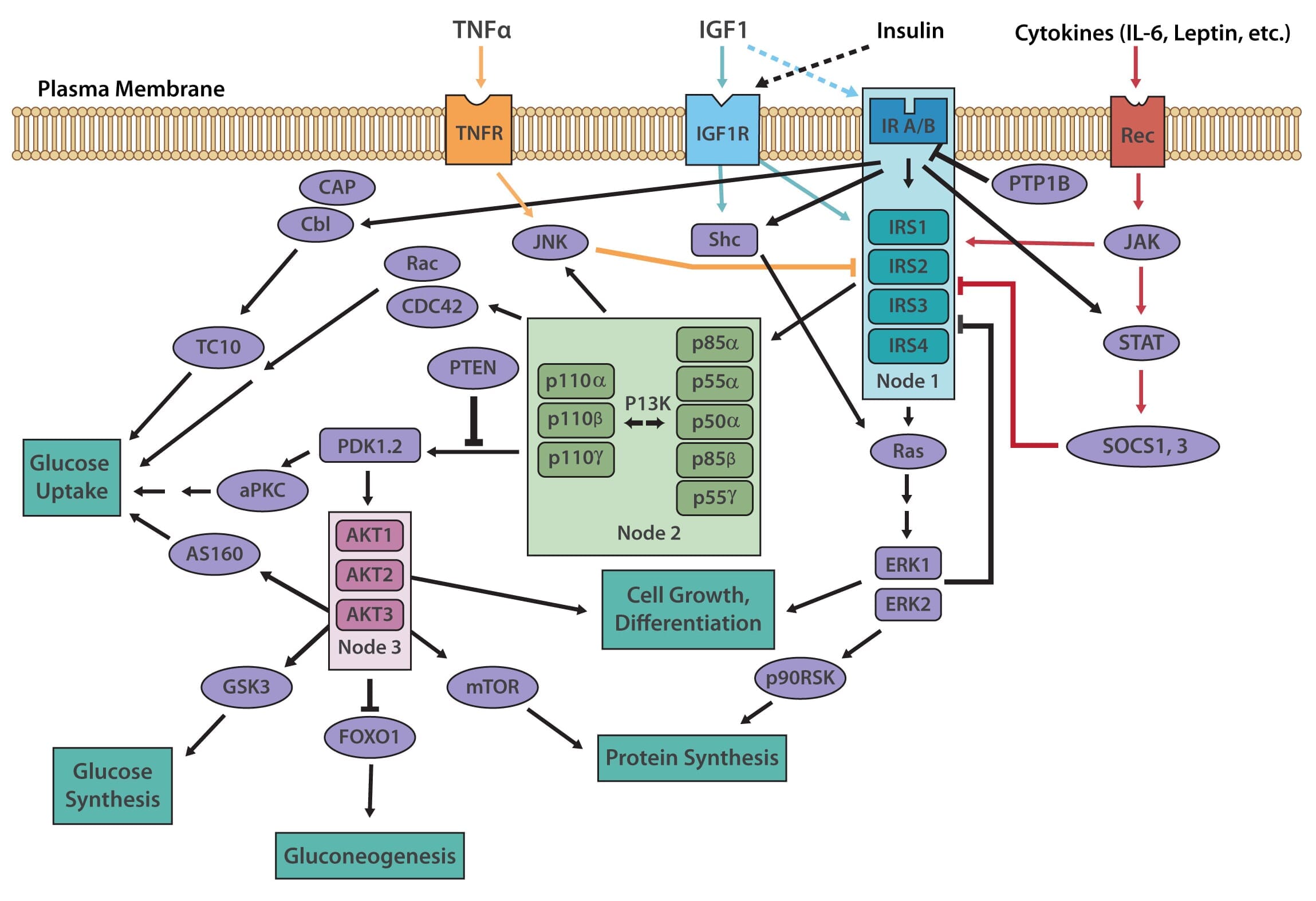
The insulin receptor belongs to the superfamily receptor tyrosine kinases (RTKs) [3,4] and is activated by insulin, as well as insulin-like growth factors (IGF1-2). It is a heterotetrametric protein consisting of two extracellular α subunits and two transmembrane β subunits, which are linked together by disulfide bonds. Most RTKs bind directly to signaling proteins. The insulin receptor, however, binds to phosphorylated residues on partner proteins, namely a family of large docking proteins known as the insulin receptor substrate family (IRS1-6) [5,6], as well as the adapter protein Shc (Src homology 2 domain containing) [7].
When insulin binds to the extracellular α subunits of the insulin receptor, a conformational change is induced, which then results in the autophosphorylation of several tyrosine residues present in the β subunits. These form the binding sites for IRS proteins, which contain phosphotyrosine (PTB) binding domains, or for Shc adapter proteins, containing src-homology 2 (SH2) domains. Binding of the insulin receptor to either IRS or Shc forms a platform that allows for the assembly of a signal transduction particle that gives rise to multiple intracellular signaling pathways [8].
Two principle pathways result from the insulin receptor-IRS interaction, the PI3K/AKT (also known as protein kinase B or PKB) pathway, and the Ras/MAPK (also known as extracellular signal regulated kinase or ERK). The PI3K (phosphoinositol 3-kinase) pathway is linked exclusively through IRS and is responsible for most of insulin's metabolic effects in the cell [9,10]. The MAPK pathway, on the other hand, stems from IRS, as well as Shc, and is involved in the regulation of gene expression and, in cooperation with the PI3K pathway, also regulates cell growth and differentiation [11].
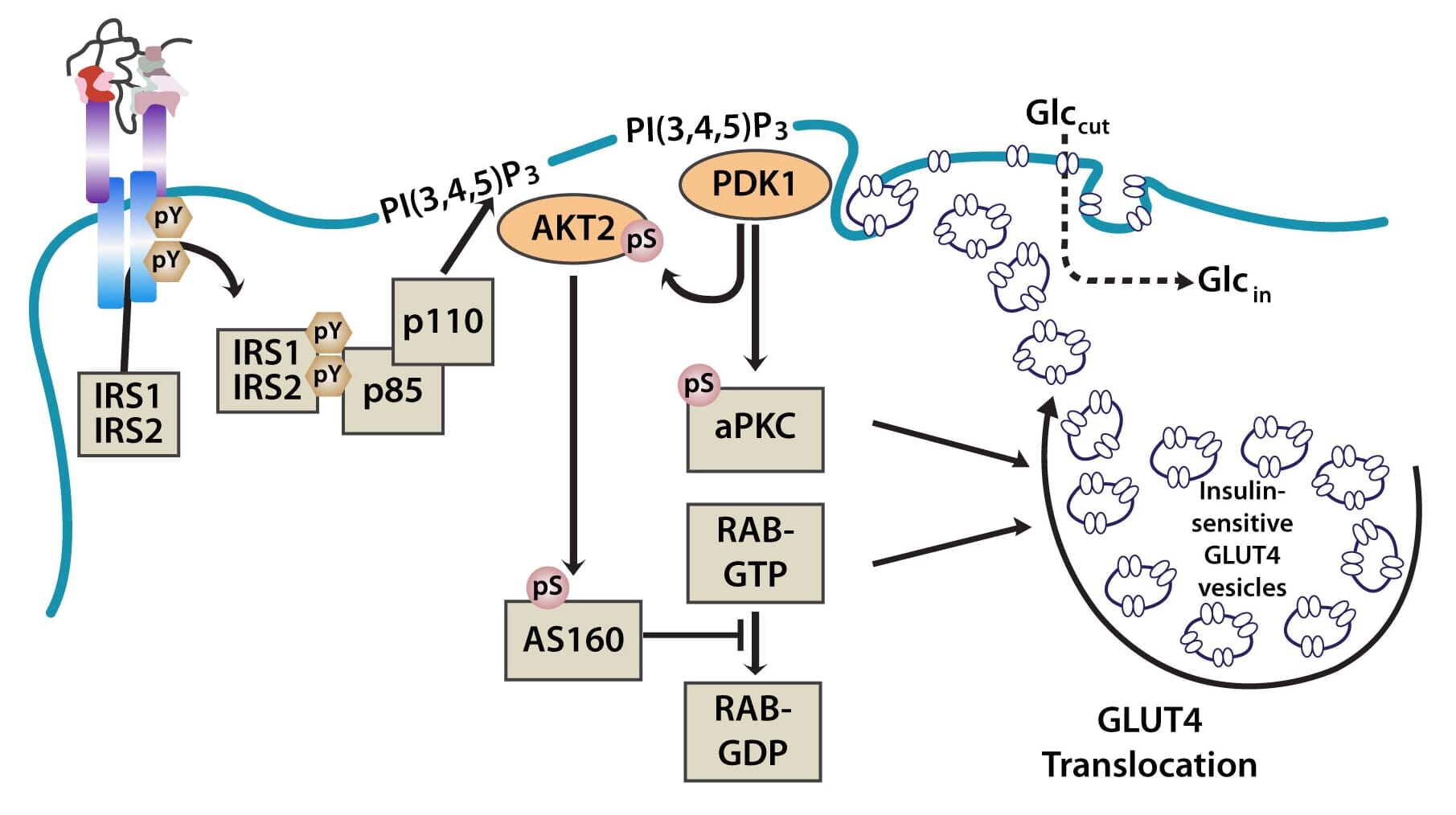
The PI3K pathway is activated by the binding of PI3K regulatory subunits p85 and p55 to IRS1 and IRS2. This results in the activation of the PI3K catalytic subunit, p110. Once the p110 subunit is activated, PI3K then catalyzes the phosphorylation of phosphatidylinositol (PI) to generate PIP3 (phosphatidylinositol 3,4,5-triphosphate) at the cell membrane [10,12]. PIP3 is an important second messenger that functions to recruit PDK1 (3-phosphoinositide dependent protein kinase-1) and AKT to the membrane, where phosphorylation of PDK1 then activates the serine/threonine residues of AKT [13]. From here, AKT plays a role in four critical downstream processes.
AKT is involved in the regulation of protein synthesis via the substrate protein mTOR, a serine/threonine kinase that functions as a nutrient sensor. mTOR stimulates protein synthesis through the phosphorylation of 4EBP1 (eukaryotic translation initiation factor 4E-binding protein 1) and p70S6K (p70 ribosomal protein S6-kinase) [14].
AKT acts in the regulation of glycogen synthesis via glycogen synthase kinase 3 (GSK3), another serine/threonine kinase, which, amongst other roles, functions to inhibit glycogen synthase. GSK3 is inhibited when phosphorylated by AKT/PKB, which results in glycogen synthesis [15].
AKT plays a role in the regulation of gluconeogenic and adipogenic genes through the transcription factor FOXO1 (forkhead box-containing protein 1, subfamily O). In the absence of insulin, FOXO1 translocates to the nucleus where it activates the expression of genes involved in gluconeogenesis, such as phosphoenolpyruvate carboxykinase (PEPCK) [13]. It also activates the expression of cyclin G2, an atypical cyclin that blocks the cell cycle, which is inhibited by insulin [16], and appears to play a key role in insulin-induced mitogenesis. When phosphorylated by AKT, FOXO1 is sequestered in the cytoplasm, and therefore cannot activate the expression of its target genes.
Importantly, AKT also regulates translocation of the insulin-sensitive glucose transporter GLUT4, which is sequestered in intracellular vesicles of muscle cells and adipocytes to the cell membrane via exocytosis, where it facilitates the uptake of glucose from the blood into cells. This is achieved through the phosphorylation of AS160 (160-kDa AKT substrate), a GTPase-activating protein that activates RAB, a small G protein involved in membrane trafficking by blocking the exchange of GTP for GDP [17].
Figure 1: The PI3K and MAPK pathways.
Figure 2: The translocation of GLUT4 in the PI3K and MAPK pathways.
The MAPK pathway is an essential secondary branch of the insulin signaling pathway. It is activated independently of the PI3K pathway either through binding of growth factor receptor-bound protein 2 (Grb2) to tyrosine-phosphorylated Shc, or through Sh2 binding to the insulin receptor. The amino-terminal SH3 domain of Grb2 binds to proline-rich regions of proteins such as son-of-sevenless (SOS), a guanine nucleotide exchange factor that catalyzes the shift of membrane-bound Ras from an inactive form (Ras-GDP) to an active form (Ras-GTP) [18]. Activated Ras-GTP is then able to stimulate downstream effectors, such as the Serine/Threonine kinase Raf, which activates its downstream targets MEK1 and MEK2 that go on to phosphorylate and activate the MAP kinases Extracellular signal-regulated kinase 1/2 (ERK1/2). Activated ERK1/2 are directly involved in multiple cellular processes, ranging from cell proliferation and differentiation. They act by regulating gene expression as well as extra-nuclear events, such as cytoskeletal reorganization, through the phosphorylation and activation of target proteins in both the cytosol and nucleus [11].
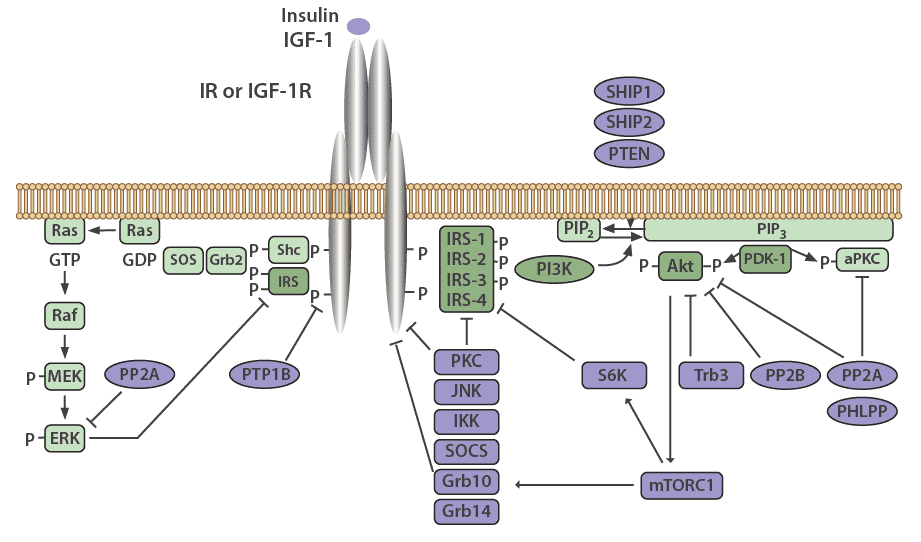
Many mechanisms exist to attenuate, finetune, and terminate insulin signaling, both at the level of the receptor and at various points in the cascade. The insulin receptor and IRS proteins are negatively regulated by multiple systems, such as ligand-induced downregulation, tyrosine protein phosphatases, and serine phosphorylation. Phosphatases also regulate the subsequent steps in the associated protein kinase cascades.
Negative feedback loops have been shown to play an essential role in finetuning this complex network [13,2]. Chronic exposure to insulin (hyperinsulinemia) results in a decrease of insulin receptors on the cell surface [19], as well as decreased IRS1 and IRS2 in vitro and in vivo in mice, which has been linked to insulin resistance in animal models [13]. The decrease in insulin receptors occurs through endocytosis by clathrin-coated vesicles. These receptors are then recycled or degraded within the lysosomes of the cell [20]. Receptor endocytosis has since been demonstrated to be a critical negative feedback mechanism that is relevant to the entire class of RTKs.
IRS signaling is negatively regulated by serine phosphorylation and kinases, such as ERK, S6 kinase, and c-Jun-N-terminal kinase (JNK), which are all activated by insulin. This is another negative feedback mechanism in the insulin signaling pathway [13]. The receptor for TNFα (TNFR), which predominantly functions in apoptosis and inflammation, induces IRS1 serine phosphorylation through JNK [13], causing insulin resistance in vitro, and in vivo in animal models as well as humans [21].
PTP1B is a major protein tyrosine phosphatase that dephosphorylates the insulin receptor. This protein resides in the endoplasmic reticulum and acts on the insulin receptor during internationalization and recycling of the receptor to the plasma membrane [22,23]. PTP1B also acts to dephosphorylate residues on activated IGF-1R and IRS proteins to reduce their activity. PTP1B knockout mice have been shown to be more sensitive to insulin and exhibit improved glucose tolerance [24,25].
The serine/threonine protein phosphatase 1 (PP1) is known to play a role in both glucose and lipid metabolism through the regulation of multiple rate-limiting enzymes, including glycogen synthase, hormone-sensitive lipase, or acetyl CoA carboxylase [26]. Protein phosphatase 2A (PP2A) also plays a critical role in regulating the activities of many protein kinases involved in the insulin cascade, including Akt, PKC, and ERK [27]. Interestingly, PP2A has been demonstrated to be hyperactivated in diabetic states [28].
Protein phosphatases 2B (PP2B), another serine/threonine phosphatase also known as calcineurin, has been shown to dephosphorylate Akt [29]. PH domain leucine-rich repeat protein phosphatases PHLPP-1 and PHLPP-2, members of the PP2C family, act to dephosphorylate both Akt and PKCs [30]. When PHLPP1 is over expressed in cells, the function of Akt and GSK3 activity is reduced. This results in a decrease in glycogen synthesis and glucose transport [31]. Obese and diabetic patients have been shown to have elevated levels of PHLPP1 in both adipose tissue and skeletal muscle which correlates with decreased Akt2 phosphorylation [31,32].
Negative regulation of the PI3K pathway occurs through dephosphorylation and subsequent inactivation of PIP3 by phospholipid phosphatases such as the tumor suppressor PTEN (phosphatase and tensin homolog) and SHIP2 (SH2-containing inositol 5'-phosphatase-2). PTEN dephosphorylates phosphoinositides on the 3'-position, whereas SHIP2 functions at the 5'-position [33].
Suppressor of Cytokine Signaling (SOCS) proteins also function to attenuate insulin receptor signaling. These are mediators of cytokine receptor signaling, such as leptin and IL-6 receptors that act through Janus kinases (JAK) and signal transduction, as well as activation of transcription (STAT) proteins [34,35]. SOCS1, SOCS3, SOCS6, and SOCS7 act by binding to the insulin receptor to inhibit signaling, as well as by targeting IRS-1 and IRS-2 for proteasomal degradation [35].
Figure 3: Negative regulators of the insulin signaling pathway.
Type 2 Diabetes
Type 2 diabetes is the primary disease associated with insulin and the insulin signaling pathways. This complex and heterogeneous disorder is caused by a combination of lifestyle and environmental factors, such as the typical western diet (which is high in fats and sugars), inactivity, and obesity, and is further modified by various genetic determinants [36]. Type 2 Diabetes is caused by two factors, insulin sensitivity (or insulin resistance) attributed to dysregulation of the insulin receptor signaling cascade, and changes in the production and secretion of insulin by the beta cells of the pancreas in response to elevated glucose. However, the relative impact of both defects on the development of diabetes has not yet been ascertained, nor have the specific molecular events at the tissue and cellular level [2]. As insulin receptors are present on many different cell types, dysregulation of the insulin signaling network effects multiple organs of the body in diabetes.
Thrombosis and Atherosclerosis
Heart attacks and strokes, precipitated by pathological blood clots (thrombi), are the leading cause of death in diabetic patients. The reason for this is twofold; firstly, patients with diabetes have an increased risk of developing more extensive atherosclerosis (AS) [37], and secondly, they possess "hyperactive" platelets, which are prone to forming thrombi. The rupture of an atherosclerotic plaque, combined with this augmented propensity for platelets to form large occlusive thrombi, increases the risk of fatal thrombotic events in diabetic individuals. Endothelial dysfunction, as well as the hyperactive phenotype of diabetic platelets, are well reported [38,39,40], but the exact underlying mechanisms remain largely unknown.
Alzheimer’s Disease
Diabetic patients also have an increased risk of developing Alzheimer's Disease (AD), a neurodegenerative disorder, although the exact relationship between these two diseases is poorly understood. Insulin signaling dysfunction has been reported in the AD brain, however, whether this is a cause or consequence of the disease has not yet been ascertained [41,42].
Cancer
There is growing evidence that abnormal insulin levels and dysregulated insulin signaling lead to cancer development and progression. A higher incidence of cancer is found in obese patients and those with type 2 diabetes. Many of the proteins that play a role in the insulin signaling pathways are involved in promoting cell proliferation and mitosis, as well as preventing apoptosis, which may increase the risk of tumor formation and metastasis [43].
Despite the tremendous progress made in understanding insulin and insulin receptor signaling over the last decades, there is still much left to be uncovered regarding how these complex networks regulate cells in both normal and disease states.
We offer a wide range of research tools that be used for studing the insulin signalling pathway, glucose storage, glucose uptake, and protein lipid synthesis through Ras, Akt, mTor and MAPK. Below we have listed some of our most popular antibodies and immunoassays.