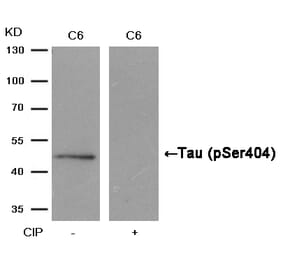
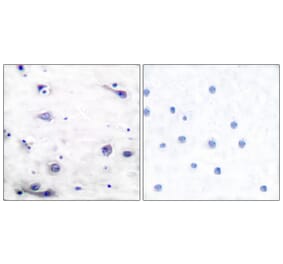
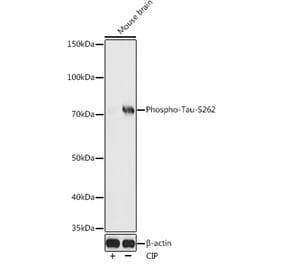
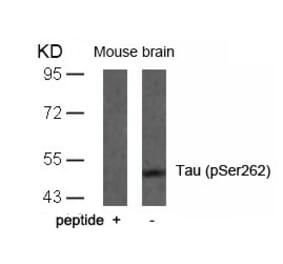
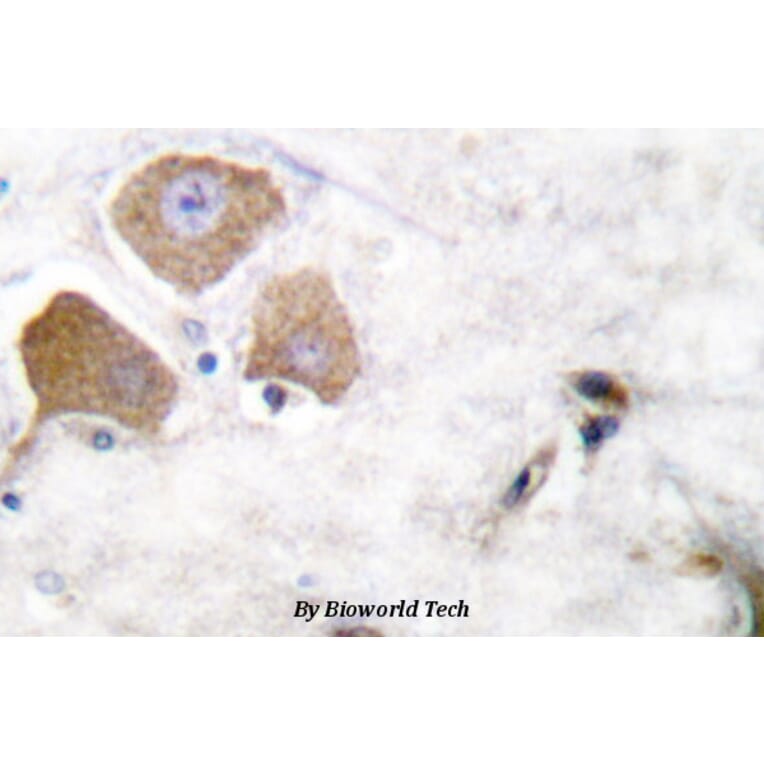
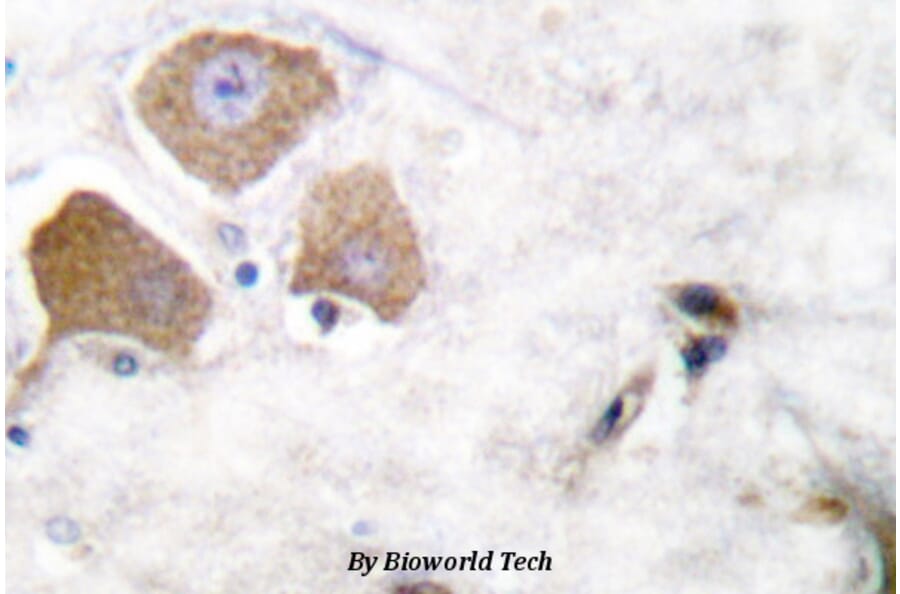
p-Tau (S396) pAb detects endogenous levels of Tau only when phosphorylated at Ser713, isoforms Tau-4 phosphorylated at Ser396, and other isoforms phosphorylated at the corresponding Site.
Applications
WB, IHC
Reactivity
Human, Mouse, Rat
Immunogen
Synthetic phosphopeptide derived from human isoforms Tau-4 around the phosphorylation site of Serine 396.
Host
Rabbit
Clonality
Polyclonal
Conjugate
Unconjugated
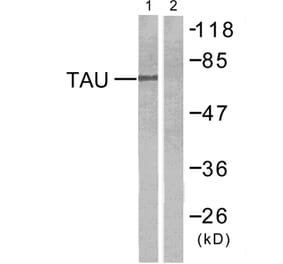
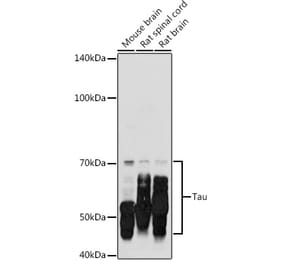
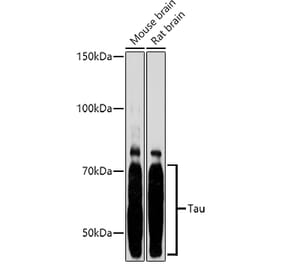
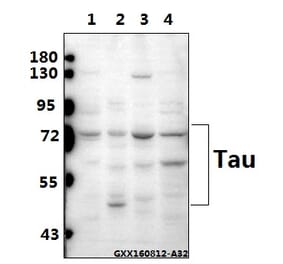
Molecular Weight
50 to 80 kDa
Purity
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen and the purity is > 95% (by SDS-PAGE).
Product Form
1 mg/ml in Phosphate buffered saline (PBS) with 0.05% sodium azide, approx. pH 7.2.


![Immunofluorescence - Anti-Tau Antibody [2E9] (A85416) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85416_1.jpg?profile=product_alternative)
![Immunofluorescence - Anti-Tau Antibody [5B10] (A85415) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85415_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-Tau (phospho Ser202 + Thr205) Antibody [AH36] (A304919) - Antibodies.com](https://cdn.antibodies.com/image/catalog/304/A304919_1.png?profile=product_alternative)


![Western Blot - Anti-Tau (phospho Thr217) Antibody [15B7] (A305063) - Antibodies.com](https://cdn.antibodies.com/image/catalog/305/A305063_1.png?profile=product_alternative)