



Primary antibodies
After blocking, samples are incubated in antibody solutions for immunolabeling. Incubation time and temperature, as well as antibody dilution and diluent should all be optimized to achieve a high SNR. All incubations are carried out in a humidified chamber – a sealed container wrapped or lined with absorbent material soaked in water. The moisture generated in the container prevents antibodies from drying out, maintaining reproducibly optimal binding conditions. Ideally the humidified chamber is also opaque or wrapped in foil to render it light-tight, important for incubations with a fluorophore-conjugated antibody.
Samples are incubated with primary antibody solutions for 3 hours at room temperature or 16 hours (overnight) at 4°C. Solutions typically include bovine serum albumin (BSA) to reduce non-specific interactions, and a detergent such as Triton X-100 or Tween-20. The best primary antibodies will detect antigens with a high degree of specificity, binding only to their target with little interaction with other cellular components. The following factors should be taken into account when selecting an antibody:
Reactivity
Antibodies recognize antigens from particular species, which will be the species that the immunizing antigen used to produce the antibody was originally from, in addition to closely related species. Antibody datasheets will typically list the species in which an antibody will recognize the antigen; this list may not be comprehensive though. For other species, researchers can empirically determine this or use protein sequence homology for reactivity prediction.
Host
The host is the species that the antibody was raised in, which is essential to know for indirection detection methods (see Direct vs indirect staining) because secondary antibodies are directed against the host species of primary antibodies, as illustrated in Figure 5. For simultaneous, indirect detection protocols (see Multicolor staining), primary antibodies should not be from the same host organism, as this will lead to cross-reactivity from secondary antibodies.
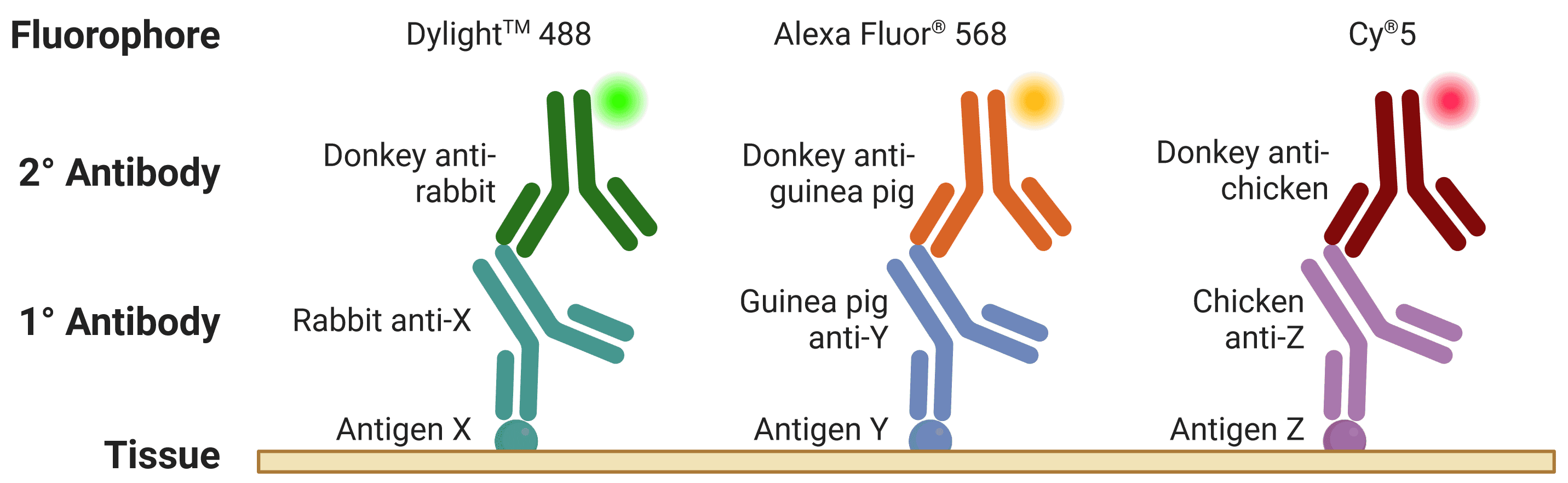
Figure 5: Multiplexed staining with ICC/IF. Multiple antigens can be detected simultaneously by using primary antibodies raised in different species. Secondary antibodies will then recognize primary antibodies from a given species, and can be resolved under a fluorescence microscope through conjugation to spectrally distinct fluorophores.
Clonality
Antibodies can be monoclonal, which target a single epitope on a target protein, or polyclonal, which recognize multiple epitopes. Monoclonal antibodies typically result in lower background signal because they are less likely to recognize non-target molecules, but they may not be robust to conformational changes in the antigen following sample fixation. In contrast, because polyclonal antibodies target multiple epitopes, some epitopes may still be accessible even after conformational change.
Polyclonal antibodies also have the advantage of being produced in a wide variety of organisms, including rat, goat, guinea pig and chicken, making multiplexing (see Multicolor staining) more feasible. Many monoclonals are available only from mice, though other species are now commercially available.
Application validity
Antibodies are validated for specific applications. Those that work for western blot will not necessarily work for ICC/IF, due to different reaction conditions and protein conformations. However, if available antibody options are limited, trialing different experimental conditions with a non-validated antibody can still yield valuable data.
Labeling and detection
Selecting the correct labeling strategy is critical to achieve immunostaining with a high SNR. Even with fully optimized fixation, permeabilization and blocking steps, use of inappropriate detection parameters can still significantly compromise results. Here we shall discuss the advantages and disadvantages of different labeling regimens, incubation conditions, and fluorophore choices to inform an experimental design that is compatible with your antigen(s) of interest and overall research objectives.
Direct vs indirect staining
Immunolabeling in ICC/IF requires the specific localization of fluorophore-conjugated antibodies to the antigen of interest with limited off-target binding events. This can be achieved through direct or indirect detection methods (Figure 6).
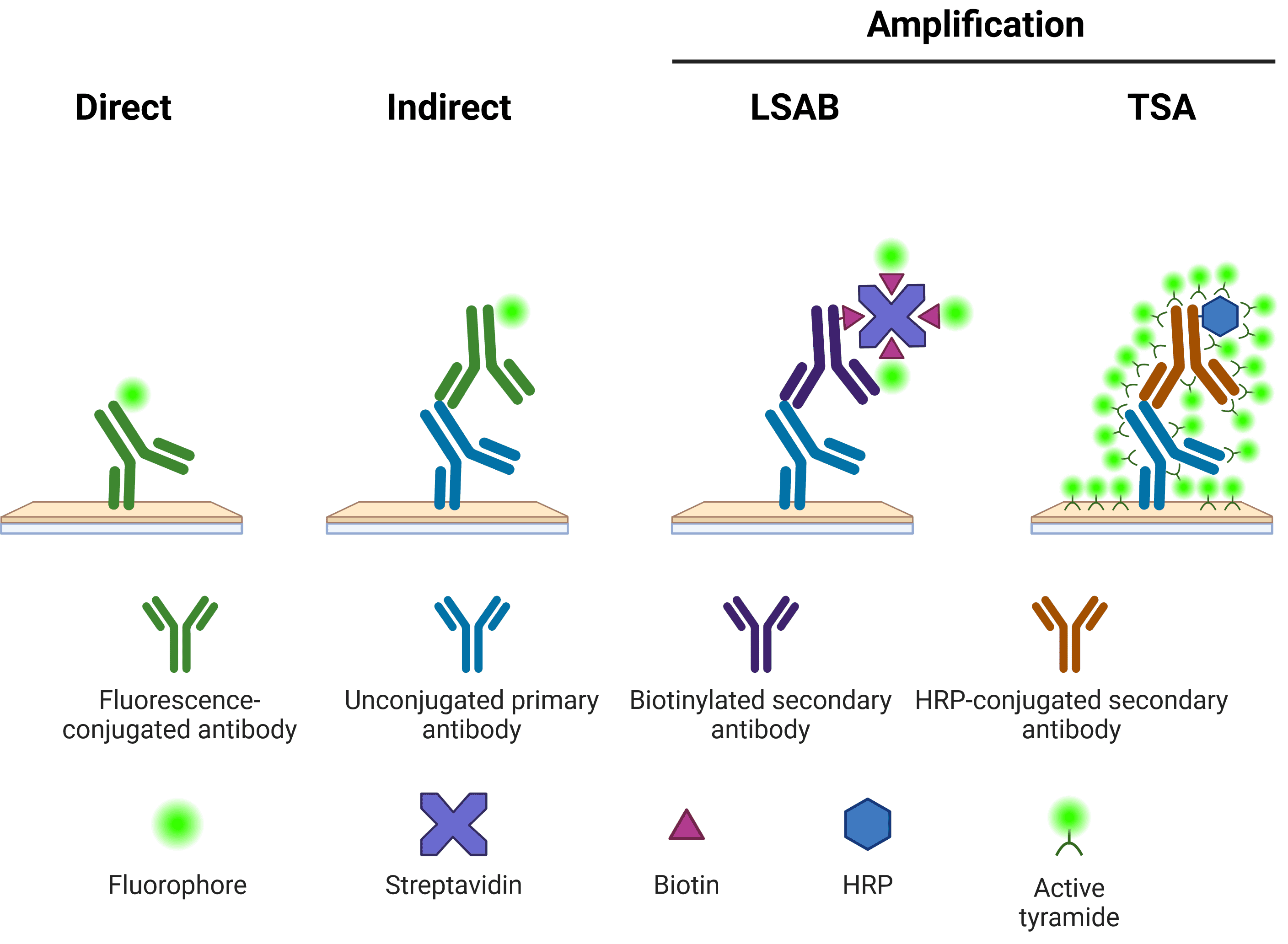
Figure 6. Schematic representation of different methods of target detection. Methods of amplification include the labeled streptavidin-biotin (LSAB) and tyramide signal amplification (TSA) systems.
Direct detection involves the use of primary antibodies directly conjugated to fluorophores for visualization of proteins in a single-step. This method is favored when dealing with high-abundance antigens where a bright, specific signal can be produced without the need for signal amplification.
Using indirect detection, the antigen-bound primary antibody is detected by a fluorophore-conjugated secondary antibody raised against the host species and isotype of the primary antibody. Since multiple secondary antibody molecules are able to bind a single primary antibody, the fluorescence signal for a single antigen detection event is amplified compared with direct methods. This approach increases the sensitivity of the assay, but it is also associated with increased background staining and care must be taken to avoid cross-reactivity between primary and secondary antibodies. Secondary antibody should be carried out in the dark using the same dilution buffer as for the primary incubation.
The advantages and disadvantages of direct and indirect detection strategies are summarized in Table 2 below:
| Direct | Indirect | |
|---|---|---|
| Time | Shorter | Longer |
| Cost | Conjugated primary antibodies are more expensive | Non-conjugated primary antibodies are cheaper, and secondary antibodies are less expensive than primary antibodies. Secondary antibodies are cost effective as they can be used with multiple primary antibodies. |
| Complexity | Single-step process | Added complexity due to use of the appropriate secondary antibody. |
| Flexibility | Experimental design limited by availability of commercial conjugated primary antibodies. Multiplexing can be easier though because primary antibodies from the same host species can be used together. | Greater variety of conjugated secondary antibodies. The same secondary antibodies can be used against multiple primary antibodies from the same host species. |
| Sensitivity | Weaker signal, but lower background. | Brighter signal due to secondary amplification. |
| Species cross-reactivity | Species cross-reactivity is minimized | Risk of secondary antibodies cross-reacting and binding primary antibodies raised in a species other than the intended target. |
Table 2:Features of direct and indirect ICC/IF.
For the detection of antigens known to be in very-low abundance, signal amplification can be enhanced further using biotin-conjugated secondary antibodies (e.g. labeled streptavidin-biotin (LSAB) system) or tyramide signal amplification (TSA). In LSAB, after binding to the primary antibody, biotinylated secondary antibodies are incubated with fluorophore-conjugated streptavidin molecules, resulting in multiple fluorophores per secondary antibody. In the TSA system, a horseradish peroxidase (HRP) enzyme is conjugated to the secondary antibody, which in turn catalyzes the conversion of labeled tyramide to a reactive radical, which binds nearby tyrosine residues. Both systems provide an extra layer of signal amplification and assay sensitivity. The trade-off with amplification systems concerns the increased processing time when performing multiple incubations in sequence.
Multicolor staining
For indirect detection of multiple colors, termed ‘multiplexing’, the immunolabeling of different antigens can be performed simultaneously or in sequence. As outlined below, simultaneous labeling involves primary and secondary antibody solutions containing the relevant antibodies for all targets. For sequential labeling, primary and secondary incubations are performed to label the first target antigen, with an intermediate blocking step before immunolabeling the second target antigen.
Simultaneous: Blocking 🠆 1st and 2nd target Primary incubation 🠆 1st and 2nd target primary incubation
Sequential: Blocking 🠆 1st target primary incubation 🠆 1st target secondary incubation 🠆 Blocking 🠆 2nd target primary incubation 🠆 2nd target secondary incubation.
Each strategy bears advantages and disadvantages. The benefits of shorter processing times using simultaneous labeling must be weighed against the risks of higher background due to incomplete removal of antibodies during wash steps and increased chance of antibody cross-reactivity. Depending on the host species the primary and secondary antibodies are raised against, sequential labeling may be required to prevent secondary antibodies cross-reacting with each other, or the wrong target primary.
Choosing fluorophores
Regardless of the labeling strategy being applied, great care must be taken to ensure the spectral properties of the chosen fluorophore(s) are compatible with the laser/filter set up of the fluorescence microscope being used for imaging. For multiplex experiments using multicolor staining, selected fluorophores must also be compatible with one another to avoid spectral overlap resulting in signal bleed-through. Commonly used fluorophores and their spectral properties are illustrated in Figure 7.
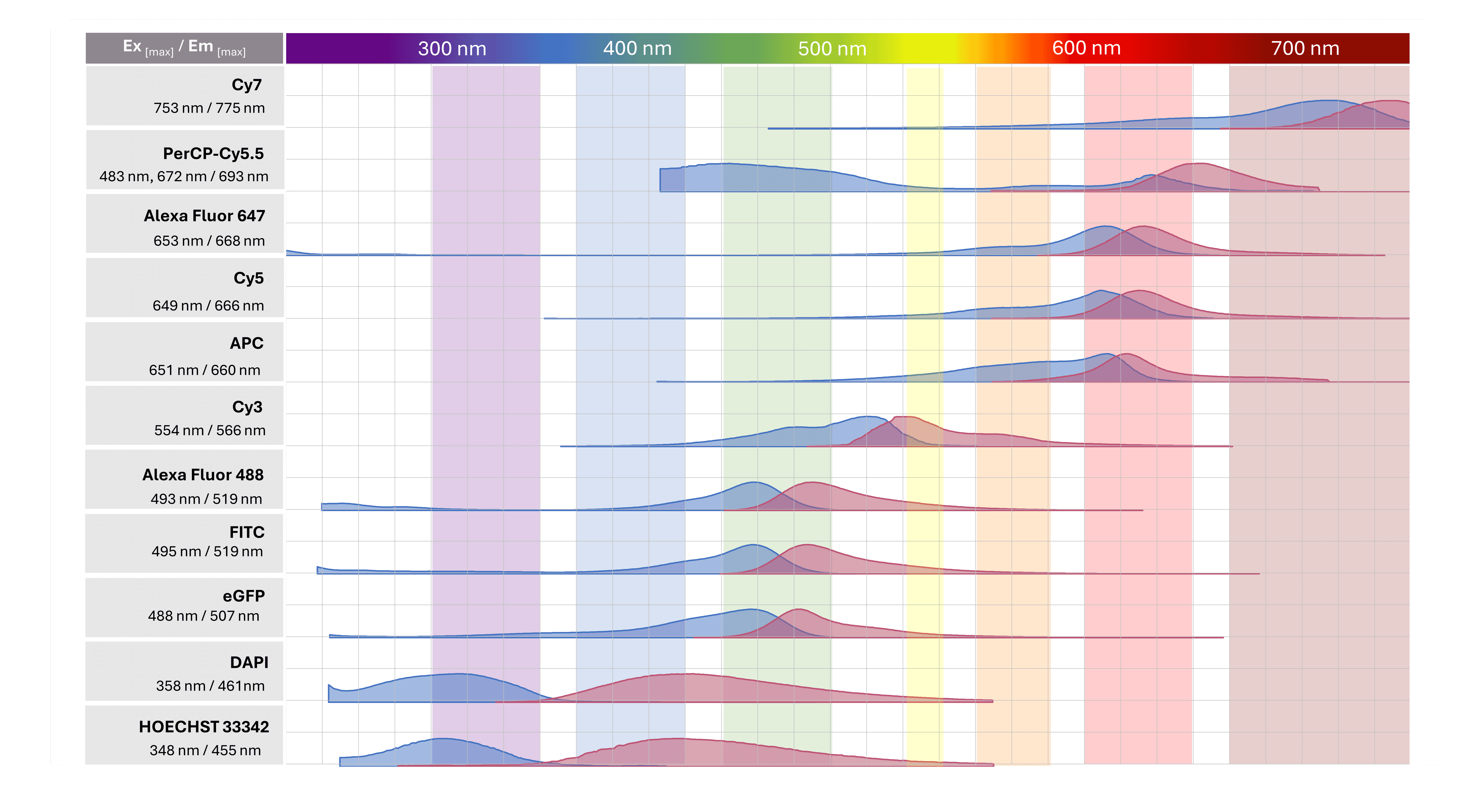
Figure 7. Commonly used fluorophores in research.
Fluorophore/Microscope compatibility
It is essential that researchers only choose fluorophores with excitation/emission spectra that are compatible with the laser and filter setup of the fluorescence microscope being used for imaging. This information can be found on the specification sheets of commercially available conjugated antibodies. Invariably, the wavelengths of light maximally absorbed during excitation and maximally emitted during fluorescence will be listed. For example, the excitation/emission maxima for TRITC fluorescent dye are 547 nm / 572 nm. While these values represent maxima, it is important to realize fluorophores still absorb and emit across a range of wavelengths, which can be visualized on freely available spectrum viewers. Depending on how broad or narrow absorption/emission profiles are, a sub-optimal laser/filter setup can still produce a dim image.
Selecting fluorophores with appropriate brightness
Intrinsic fluorophore brightness is determined by two properties, the extinction coefficient (ε) and the quantum yield (ϕ). The extinction coefficient is a measure of absorption efficiency, providing the probability that a fluorophore will absorb a photon of light at a given wavelength. The quantum yield measures emission efficiency, a ratio reporting the average number of photons emitted of a given wavelength for every photon absorbed, also of a given wavelength. These properties can be used to distinguish bright dyes from those that are dim.
It is helpful to consider the brightness of a chosen fluorophore in the context of target antigen abundance. The brightness of the detectable signal should not be excessive, thus specific and sensitive antibodies targeting abundant antigens should be conjugated to fluorophores with lower brightness. Equally, the signal produced detecting low-abundance antigens can be boosted through the use of bright fluorophores with high ε and ϕ values. Managing signal brightness is particularly useful when performing multi-color experiments. Complementing fluorophore selection against differing antigen abundance will ensure similar signal brightness between targets upon imaging. This simplifies microscope setup with even, minimal exposure times that minimize photobleaching.
Photobleaching
Photobleaching is a phenomenon which diminishes fluorophore fluorescence following prolonged exposure to excitation radiation. This can be minimized through the selection of fluorophores which are more photostable. In addition to choosing fluorophores with high photostability, photobleaching can be controlled by limiting the intensity and duration of excitation by decreasing laser power and reducing exposure times respectively. Use of mounting media with anti-fade reagents also minimizes photobleaching effects in samples.
Spectral overlap (bleed-through)
Each fluorophore emits radiation across a range of wavelengths; this constitutes its emission spectral profile. During multi-color experiments, it is important that combinations of fluorophores are used which are as isolated as possible (Figure 8). This means that spectral overlap is minimized, ensuring the detected signal through one filter has not been emitted by the fluorophore belonging to a neighboring filter set. When designing experiments pay close attention to the spectral properties of fluorescent probes and dyes being used; this includes the emission profiles of nuclear stains, for example, DAPI. In addition to careful fluorophore choice, emission filter thresholds on microscopes can also help to mitigate bleed-through.
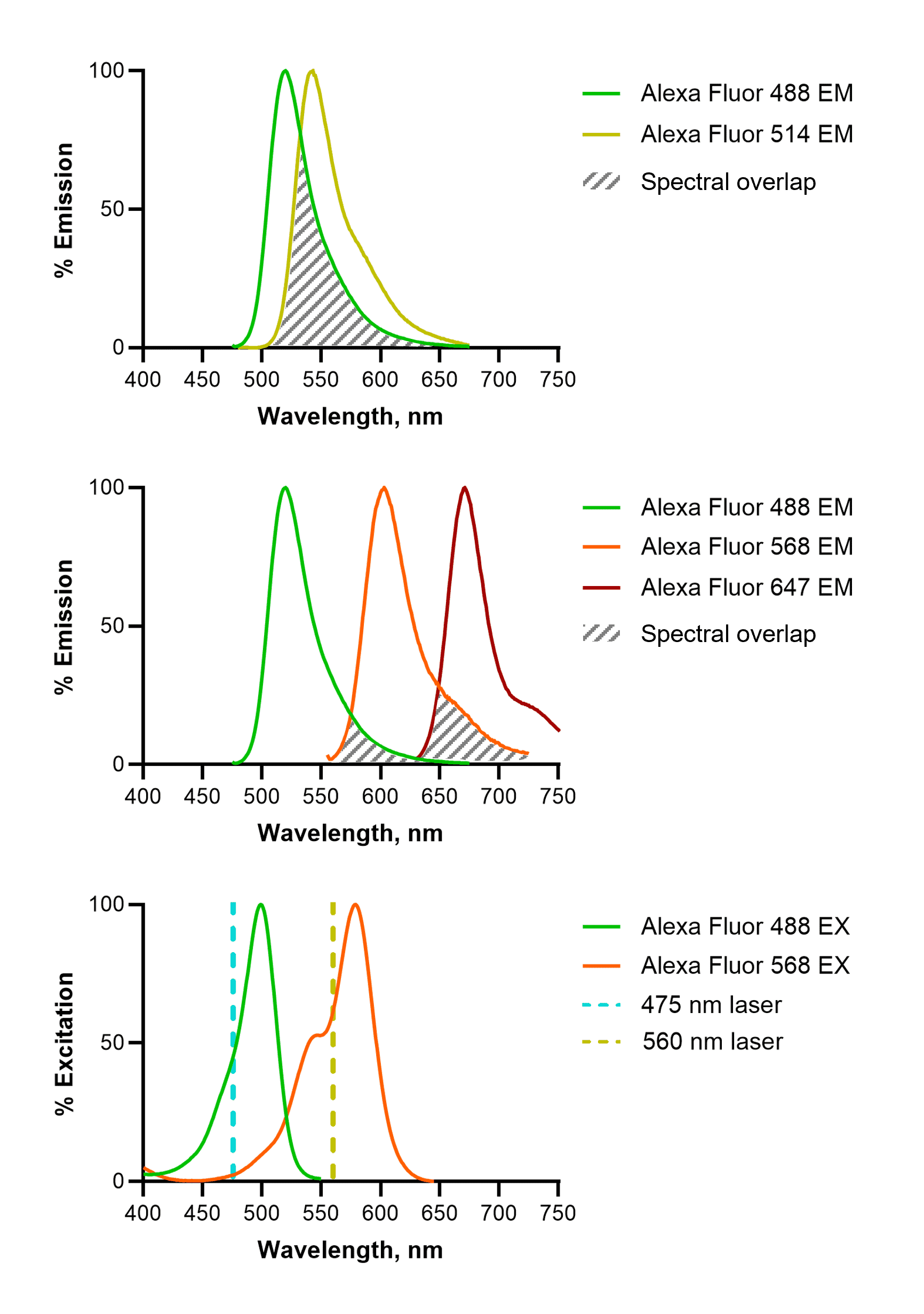
Figure 8: Spectral overlap in fluorescence imaging. Fluorophores that emit in similar spectral ranges, such as Alexa Fluor (AF)488 and AF514 are difficult or impossible to distinguish (top). This can be mitigated by choosing well-spaced fluorophores (middle), and by choosing fluorophores that are activated by distinct lasers (bottom). For example, AF568 shows almost no excitation by a 475 nm laser, which is required to excite AF488. EM: emission; EX: excitation.
Counterstaining and mounting
Following detection of the target antigens by immunolabeling samples can be counterstained in order to define individual cells and cell structures. The most common fluorescent dyes delineate nuclei by binding DNA, such as DAPI (0.1 μg / ml) or Hoechst 33342 (0.1 – 1 μg / ml). Nuclear counterstains are typically incubated with samples at room temperature for 5 - 15 minutes, or may even be incorporated into the mounting medium.
Non-nuclear regions can also be identified, such as by using phalloidin (200 units/ml or 6.6 μM), an F-actin-binding fluorescent dye that highlights the cytoplasm. Such staining often takes place at the same time as primary antibody incubations.
Like fluorophores conjugated to the secondary antibodies, counterstains have inherent spectral properties that must not overlap with other fluorophores being used. For example, DAPI emits strongly in the blue region of the spectrum, thus should not be used alongside blue dyes such as Alexa Fluor 405.
Table 3 below lists some common fluorescent dyes for use in ICC/IF. A number of fluorescent dyes for other organelles, such as mitochondria and Golgi, are also available commercially for both live and fixed cell imaging.
| Name | Target | Color |
|---|---|---|
| DAPI | Nucleic acids | Blue |
| Hoechst 33342 | Nucleic acids | Blue |
| Hoechst S769121 | Nucleic acids | Yellow |
| DRAQ5 | Nucleic acids | Far red |
| DRAQ7 | Nucleic acids | Far red |
| Phalloidin | Actin filaments | Conjugated to various fluorophores |
| Wheat germ agglutinin (WGA) | Plasma membrane | Conjugated to various fluorophores |
Table 3:Commonly used fluorescent counterstains and their target organelle.
After counterstaining, coverslips are ready to be mounted on to glass slides for storage and imaging. A drop of mounting medium is added to each slide before the coverslips are gently lowered onto the slide with the cells facing down. Care should be taken to avoid the formation of air bubbles between the slide and coverslip. Depending on the mounting medium being used, the perimeter of the coverslips may need to be sealed using clear nail varnish. This is not required for hard-setting media, which should instead be left to dry and polymerize fully, sometimes for as long as 24 hours. For improved detection sensitivity with dyes which have low photostability and rapidly bleach, anti-fade mounting media products should be preferred. Samples can be stored in the dark at 4°C for several months before imaging depending on the stability of the fluorophores.
Sample visualization
Once prepared, samples can be visualized by fluorescence microscopy. Excitation radiation is emitted by a light source before passing through an excitation filter. Possible light sources include mercury-vapor lamp, LEDs, and lasers. Filtered excitation radiation is reflected on to the sample by a dichroic mirror where it is absorbed by fluorophores. Emitted radiation is transmitted through the dichroic mirror before passing through an emission filter. Fluorescence signal is viewed through the eye-piece or detected by a monochrome camera. Confocal microscopy utilizes ‘optical sectioning’ to improve the spatial resolution of the fluorescence image, using a pinhole to exclude light from outside the focal plane. Other setups allow for rapid, quantitative single-cell analyses to be performed through high-content screening of multiple samples.
References
Diagrams created with BioRender.com.
- Wu, Y., Li, Q. & Chen, X.-Z. Detecting protein–protein interactions by far western blotting. Nat. Protoc. 2, 3278–3284 (2007).
- Arkin, M. R., Glicksman, M. A., Fu, H., Havel, J. J. & Du, Y. Inhibition of Protein-Protein Interactions: Non-Cellular Assay Formats. in Assay Guidance Manual (2012).
- Behring, E. A. von & Kitasato, S. Ueber das Zustandekommen der Diphtherie-Immunität und der Tetanus-Immunität bei Thieren. Dtsch. Med. Wochenschr. 16, 1113–1114 (1890).
- Behring, E. A. von. Untersuchungen über das Zustandekommen der Diphtherie-Immunität bei Thieren. DMW - Dtsch. Med. Wochenschr. 16, 1145–1148 (1890).
- Holyoak, T. Molecular Recognition: Lock-and-Key, Induced Fit, and Conformational Selection. in Encyclopedia of Biophysics, 1584–1588 (2013). doi:10.1007/978-3-642-16712-6_468.
- The Nobel Prize in Physiology or Medicine 1908. NobelPrize.org.
- Acuña, A. U. & Amat-Guerri, F. Early History of Solution Fluorescence: The Lignum nephriticum of Nicolás Monardes. in Fluorescence of Supermolecules, Polymers, and Nanosystems 3–20 (2008).
- Stokes, G. G. XXX. On the change of refrangibility of light. Philos. Trans. R. Soc. Lond. 142, 463–562 (1852).
- Baeyer, A. Ueber eine neue Klasse von Farbstoffen. Berichte Dtsch. Chem. Ges. 4, 555–558 (1871).
- Coons, A. H., Creech, H. J. & Jones, R. N. Immunological Properties of an Antibody Containing a Fluorescent Group. Proc. Soc. Exp. Biol. Med. 47, 200–202 (1941).
- Riggs, J. L., Seiwald, R. J., Burckhalter, J. H., Downs, C. M. & Metcalf, T. G. Isothiocyanate Compounds as Fluorescent Labeling Agents for Immune Serum. Am. J. Pathol. 34, 1081–1097 (1958).
- Southwick, P. L. et al. Cyanine dye labeling reagents—carboxymethylindocyanine succinimidyl esters. Cytometry 11, 418–430 (1990).
- Renz, M. Fluorescence microscopy—A historical and technical perspective. Cytometry A 83, 767–779 (2013).
- Ploem, J. S. The use of a vertical illuminator with interchangeable dichroic mirrors for fluorescence microscopy with incidental light. Z. Wiss. Mikrosk. Mikrosk. Tech. 68, 129–142 (1967).
- Leavy, O. The birth of monoclonal antibodies. Nat. Immunol. 17, S13–S13 (2016).
- Griffiths, G. Introduction to Immunocytochemistry and Historical Background. in Fine Structure Immunocytochemistry, 1–8 (1993).
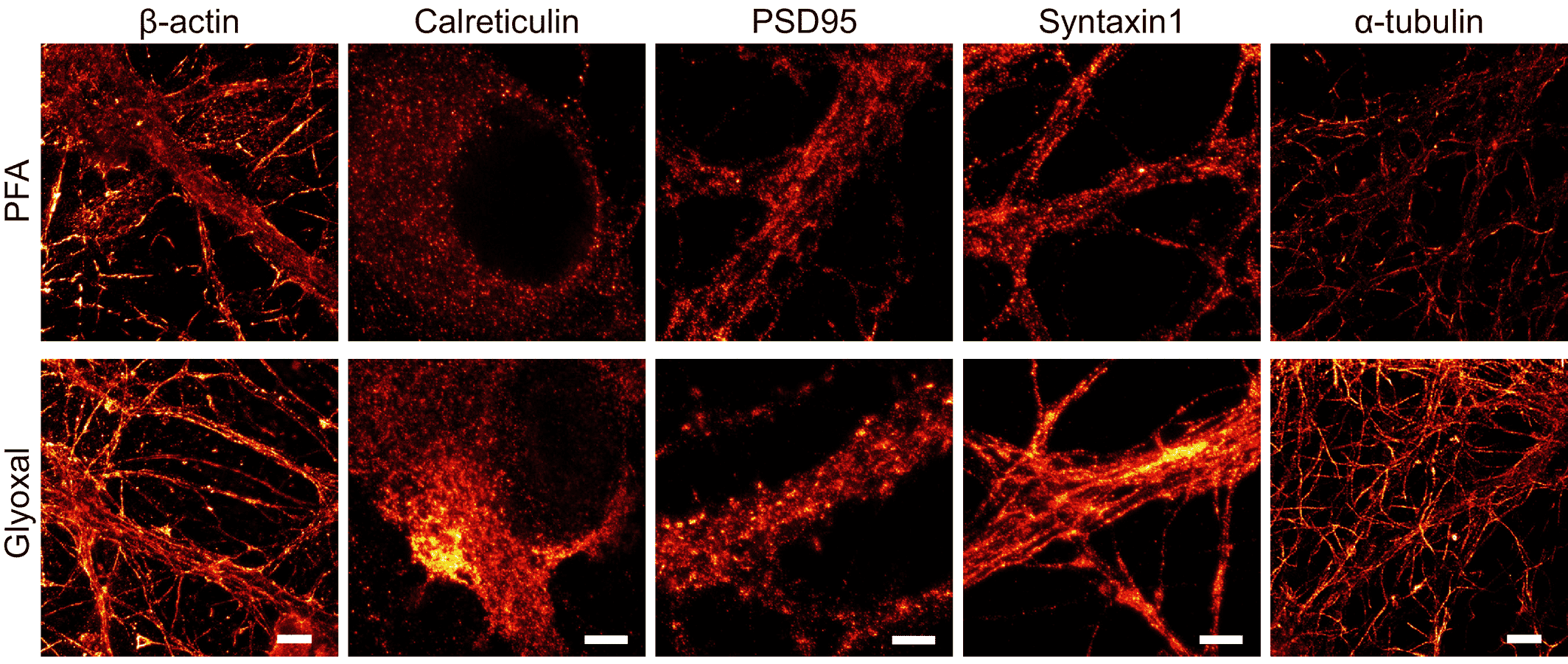
- Richter, K. N. et al. Glyoxal as an alternative fixative to formaldehyde in immunostaining and super‐resolution microscopy. EMBO J. 37, 139–159 (2018).
- Konno, K., Yamasaki, M., Miyazaki, T. & Watanabe, M. Glyoxal fixation: An approach to solve immunohistochemical problem in neuroscience research. Sci. Adv. 9, eadf7084 (2023).