Unconjugated
Parkinson's Disease (PD) is primarily characterized by a-synuclein pathology, which manifests as intraneuronal inclusions, neuroinflammation, and neurodegeneration. However, emerging evidence also points to significant vascular impairments as a critical aspect of PD pathology, which remains largely underexplored due to the inability of traditional in vitro models to recapitulate such vascular changes. To address this unmet need, here we combine the human organ-on-a-chip technology with the principle of vasculogenic self-assembly to engineer the capillary interface of dopaminergic neurons in the substantia nigra pars compacta of the midbrain. In our proof-of-concept demonstration, we successfully recreated critical neuronal pathology in PD, including a-synuclein aggregation, inflammatory responses, and progressive neuronal degeneration, by exposing our model to specially generated PD-associated a-synuclein preformed fibrils. Importantly, this engineering approach also enables the investigation of progressive vascular changes characteristic of PD, such as endothelial dysfunction, barrier disruption, and vascular regression. Our sophisticated PD model establishes a novel platform for exploring the multifaceted nature of the disease and understanding the complex interplay between neurodegeneration and vascular pathology, offering a unique tool for developing innovative therapeutic strategies that address both the neuronal and vascular components of PD pathology.
Alzheimer's disease (AD) is characterized by the accumulation of soluble amyloid-ß oligomers (AßOs) in the brain, which disrupt synaptic function and promote cognitive decline. Here, we investigated the effects of AßOs on excitatory and inhibitory synaptic transmission and plasticity by performing stereotaxic injections of AßOs directly into the hippocampal CA1 region, followed by hippocampal slice isolation for electrophysiological measurements. AßOs injections altered basal excitatory synaptic transmission, reducing field excitatory postsynaptic potentials (fEPSPs) and impairing excitatory long-term potentiation (LTP). Additionally, AßOs injections significantly increased basal inhibitory synaptic transmission, as evidenced by the increased amplitude of field inhibitory postsynaptic potentials (fIPSPs), but impaired the induction and maintenance of inhibitory long-term potentiation (iLTP). Accordingly, we propose that AßOs injections induce the saturation of the GABAergic system and thus disrupt the hippocampal excitatory-inhibitory balance. These findings highlight the dual impact of AßOs on both excitatory and inhibitory synapses, generating synaptic dysregulation and possibly worsening cognitive decline in AD. Understanding these mechanisms could provide new insights for developing therapies to restore synaptic balance and hippocampal function in AD.


![Immunofluorescence - Anti-MAP2 Antibody [5H11] (A85296) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85296_1.jpg?profile=product_alternative)
![Immunofluorescence - Anti-MAP2 Antibody [4H5] (A85297) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85297_1.jpg?profile=product_alternative)
![Immunofluorescence - Anti-MAP2 Antibody [2C4] (A85459) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85459_1.jpg?profile=product_alternative)
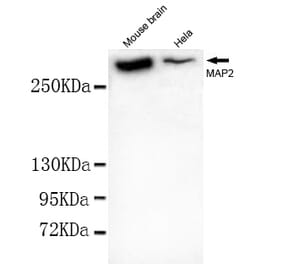
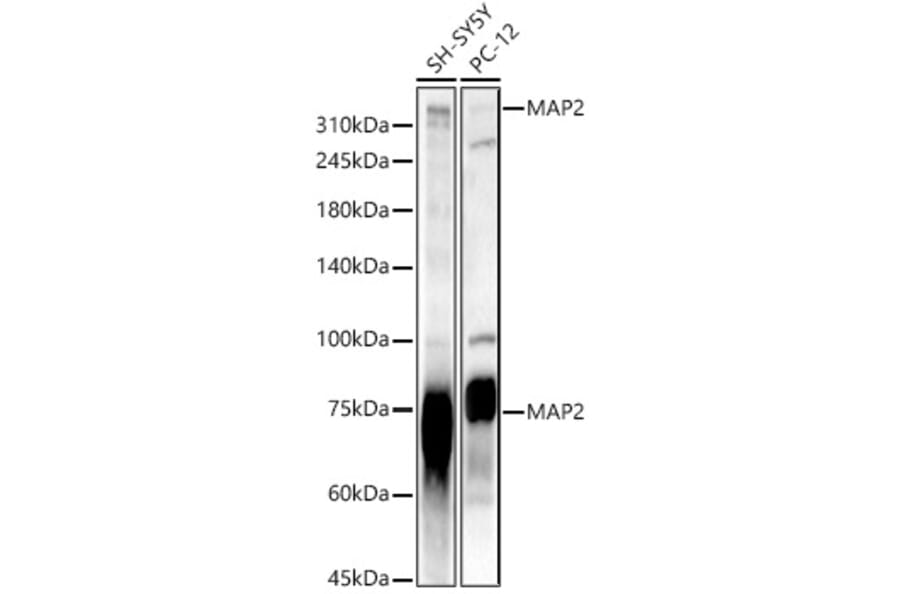
![WB - Anti-MAP2 Antibody [MT-07] (A86618)](https://cdn.antibodies.com/image/catalog/86/A86618_1.jpg?profile=product_alternative)
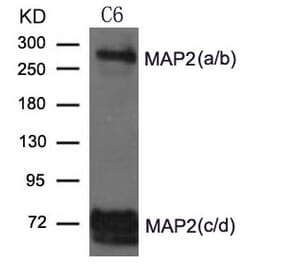
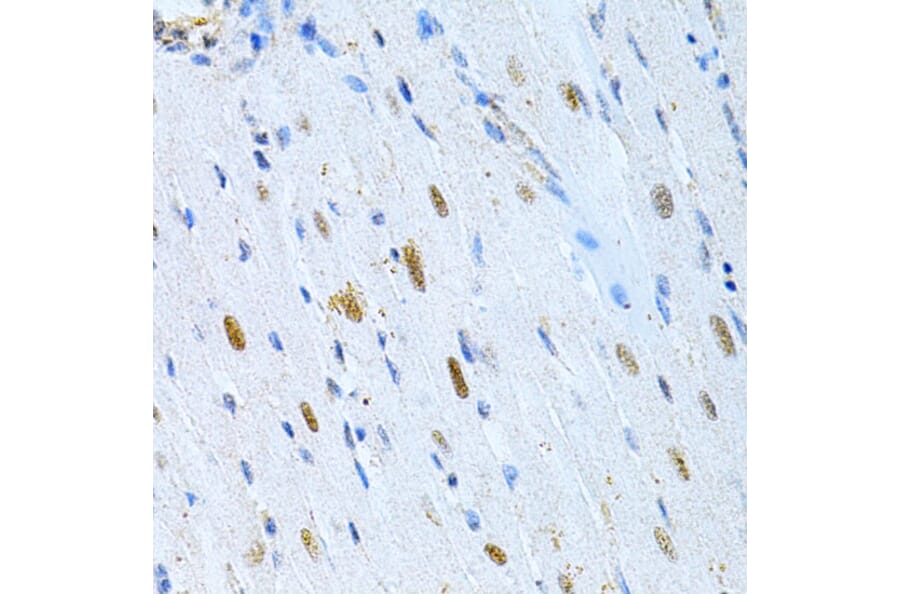
![WB - Anti-MAP2 Antibody [MT-08] (A86619)](https://cdn.antibodies.com/image/catalog/86/A86619_1.jpg?profile=product_alternative)
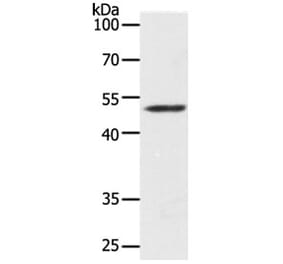
![WB - Anti-MAP2 Antibody [MT-01] (A86786)](https://cdn.antibodies.com/image/catalog/86/A86786_1.jpg?profile=product_alternative)