Unconjugated
Bromodomain containing 7 (BRD7) was identified as a nuclear transcriptional regulatory factor. BRD7 functions as a tumor suppressor in multiple cancers, including nasopharyngeal carcinoma (NPC). In this study, we reported a novel mechanism of BRD7 in NPC progression. We demonstrated that the expression of miR-141 was remarkably increased in NPC tissues and was negatively correlated with the expression of BRD7 and the survival rate of NPC patients. Decreased expression levels of miR-141, including the primary, the precursor and the mature forms of miR-141, were found in BRD7-overexpressing HEK293, 5-8F and HNE1 cells compared the control cells, while there was no obvious effect on the expression levels of the two critical enzymes Drosha and Dicer. BRD7 can negatively regulate the promoter activity of miR-141, while no obvious binding site of BRD7 was found in the potential promoter region of miR-141. Moreover, ectopic expression of miR-141 can significantly promote cell proliferation and inhibit apoptosis in NPC, and rescuing the expression of miR-141 in BRD7-overexpressing NPC cells could partially reverse the tumor suppressive effect of BRD7 on cell proliferation and tumor growth in vitro and in vivo. Furthermore, the activation of the PTEN/AKT pathway mediated by the overexpression of BRD7 could be inhibited by rescuing the expression of miR-141, which accordingly results in the partial restoration of cell proliferation and tumor growth. Our findings demonstrate that the BRD7/miR-141/PTEN/AKT axis has critical roles in the progression of NPC and provide some promising targets for the diagnosis and treatment of NPC.
Clinical studies have shown hyperuricemia strongly associated with insulin resistance as well as cardiovascular disease. Direct evidence of how high uric acid (HUA) affects insulin resistance in cardiomyocytes, but the pathological mechanism of HUA associated with cardiovascular disease remains to be clarified. We aimed to examine the effect of HUA on insulin sensitivity in cardiomyocytes and on insulin resistance in hyperuricemic mouse model. We exposed primary cardiomyocytes and a rat cardiomyocyte cell line, H9c2 cardiomyocytes, to HUA, then quantified glucose uptake with a fluorescent glucose analog, 2-NBDG, after insulin challenge and detected reactive oxygen species (ROS) production. Western blot analysis was used to examine the levels of insulin receptor (IR), phosphorylated insulin receptor substrate 1 (IRS1, Ser307) and phospho-Akt (Ser473). We monitored the impact of HUA on insulin resistance, insulin signaling and IR, phospho-IRS1 (Ser307) and phospho-Akt levels in myocardial tissue of an acute hyperuricemia mouse model established by potassium oxonate treatment. HUA inhibited insulin-induced glucose uptake in H9c2 and primary cardiomyocytes. It increased ROS production; pretreatment with N-acetyl-L-cysteine (NAC), a ROS scavenger, reversed HUA-inhibited glucose uptake induced by insulin. HUA exposure directly increased the phospho-IRS1 (Ser307) response to insulin and inhibited that of phospho-Akt in H9C2 cardiomyocytes, which was blocked by NAC. Furthermore, the acute hyperuricemic mice model showed impaired glucose tolerance and insulin tolerance accompanied by increased phospho-IRS1 (Ser307) and inhibited phospho-Akt response to insulin in myocardial tissues. HUA inhibited insulin signaling and induced insulin resistance in cardiomyocytes in vitro and in vivo, which is a novel potential mechanism of hyperuricemic-related cardiovascular disease.
Triple-negative breast cancer (TNBC) patients have poor prognosis due to the aggressive metastatic behaviors. Our study reveals that expression of estrogen related receptor α (ERRα) is significantly (p < 0.01) positively associated with high grade tumors and lymph node metastasis, while negatively correlated with overall survival (OS), in 138 TNBC patients. Targeted inhibition of ERRα by its inverse agonist XCT-790 or si-RNA obviously inhibits in vitro motility of TNBC cells. While over expression of ERRα triggers the invasion and migration of TNBC cells. Further, si-ERRα and XCT-790 inhibit the epithelial mesenchymal transition (EMT) of TNBC cells with increasing the expression of E-cadherin and decreasing fibronectin (FN) and vimentin. While XCT-790 has no effect on the expression of EMT related transcription factors such as Snail or Slug. Further, inhibitors of MAPK, PI3K/Akt, NF-κB signal molecules, which are activated by XCT-790, can not attenuate the suppression effects of XCT-790 on EMT. Alternatively, luciferase reporter gene assays and ChIP analysis indicate that ERRα can directly bind with FN promoter at ERR response element-3 (ERRE-1), ERRE-3, and ERRE-4, while XCT-790 reduces this bond. In vivo data show that ERRα expression is significantly (p < 0.05) correlated with FN in clinical TNBC patients. In MDA-MB-231 tumor xenograft models, XCT-790 decreases the expression of FN, inhibits the growth and lung metastasis, and suppresses the EMT. Our results demonstrate that ERRα functions as a metastasis stimulator and its targeted inhibition may be a new therapeutic strategy for TNBC treatment.
Metastasis, the greatest clinical challenge associated with cancer, is closely connected to multiple biological processes, including invasion and adhesion. The hypoxic environment in tumors is an important factor that causes tumor metastasis by activating HIF-1α. Fucoidan, extracted from brown algae, is a sulfated polysaccharide and, as a novel marine biological material, has been used to treat various disorders in China, Korea, Japan and other countries. In the present study, we demonstrated that fucoidan derived from Undaria pinnatifida sporophylls significantly inhibits the hypoxia-induced expression, nuclear translocation and activity of HIF-1α, the synthesis and secretion of VEGF-C and HGF, cell invasion and lymphatic metastasis in a mouse hepatocarcinoma Hca-F cell line. Fucoidan also suppressed lymphangiogenesis in vitro and in vivo. In addition, accompanied by a reduction in the HIF-1α nuclear translocation and activity, fucoidan significantly reduced the levels of p-PI3K, p-Akt, p-mTOR, p-ERK, NF-κB, MMP-2 and MMP-9, but increased TIMP-1 levels. These results indicate strongly that the anti-metastasis and anti-lymphangiogenesis activities of fucoidan are mediated by suppressing HIF-1α/VEGF-C, which attenuates the PI3K/Akt/mTOR signaling pathways.
The protein tyrosine phosphatase 1B (PTP1B), a non-transmembrane protein tyrosine phosphatase, has been implicated in gastric pathogenesis. Several lines of recent evidences have shown that PTP1B is highly amplified in breast and prostate cancers. The aim of this study was to investigate PTP1B amplification in gastric cancer and its association with poor prognosis of gastric cancer patients, and further determine the role of PTP1B in gastric tumorigenesis. Our data demonstrated that PTP1B was significantly up-regulated in gastric cancer tissues as compared with matched normal gastric tissues by using quantitative RT-PCR (qRT-PCR) assay. In addition, copy number analysis showed that PTP1B was amplified in 68/131 (51.9%) gastric cancer cases, whereas no amplification was found in the control subjects. Notably, PTP1B amplification was positively associated with its protein expression, and was significantly related to poor survival of gastric cancer patients. Knocking down PTP1B expression in gastric cancer cells significantly inhibited cell proliferation, colony formation, migration and invasion, and induced cell cycle arrested and apoptosis. Mechanically, PTP1B promotes gastric cancer cell proliferation, survival and invasiveness through modulating Src-related signaling pathways, such as Src/Ras/MAPK and Src/phosphatidylinositol-3-kinase (PI3K)/Akt pathways. Collectively, our data demonstrated frequent overexpression and amplification PTP1B in gastric cancer, and further determined the oncogenic role of PTP1B in gastric carcinogenesis. Importantly, PTP1B amplification predicts poor survival of gastric cancer patients.
The goal of this study was to investigate the possible protective effects of sitagliptin against dyslipidemia-related kidney injury in apolipoprotein E knockout (apoE-/-) mice. Eight-week-old male apoE-/- mice were randomized to receive either a high fat diet (HFD, apoE-/- group) or HFD mixed with sitagliptin (sita + apoE-/- group) for 16 weeks. A control group of age- and gender-matched C57BL/6J mice were fed a HFD. The apoE-/- group exhibited increases in body weight and serum lipid levels in addition to high-density lipoprotein, and increases in 24-h urinary 8-hydroxy-2-deoxyguanosine and albuminuria excretion. Decreased insulin sensitivity was also observed in the apoE-/- group. These mice additionally contained enlargements of the glomerular mesangial matrix area, lipid deposition area, and renal interstitium collagen area. The apoE-/- group also demonstrated down-regulation of phosphorylated AMP-activated protein kinase (AMPK), increases in renal mRNA expression of transforming growth factor-beta 1 (TGF-β1) and fibronectin (FN), and increased protein expression of Akt, TGF-β1, FN and p38/ERK mitogen-activated protein kinase (MAPK). Sitagliptin treatment successfully ameliorated all the deleterious effects of dyslipidemia tested. To our knowledge, this is the first time that sitagliptin has been shown to reverse the renal dysfunction and structural damage induced by dyslipidemia in apoE-/- mice. Our results suggest that the renoprotective mechanism of sitagliptin may be due to a reduction in Akt levels, a restoration of AMPK activity, and inhibition of TGF-β1, FN, and p38/ERK MAPK signaling pathways.
The expression and function of P-glycoprotein (P-gp) is associated with the phenotype of multi-drug resistance (MDR), leading chemotherapy failure of patients suffered with cancer. Grape seed procyanidin(GSP) is a natural polyphenol supplement with anti-inflammatory effect. Present study assessed a new use of GSP on the MDR reversal activity and its possible molecular mechanisms in MDR1-overpressing paclitaxel resistant ovarian cancer cells. Our results showed GSP significantly enhanced the cytotoxicity of paclitaxel and adriamycin in paclitaxel resistant A2780/T cells but its parental A2780 cells. Furthermore, GSP strongly inhibited P-gp expression by blocking MDR1 gene transcription, as well as, increased the intracellular accumulation of the P-gp substrate rhodamine-123 in A2780/T cells. Nuclear factor-κB(NF-κB) activity, IκB degradation level and NF-κB/p65 nuclear translocation induced by lipopolysaccharide (LPS) and receptor activator for nuclear factor-κB ligand (RANKL) were markedly inhibited by pre-treatment with GSP. Meanwhile, GSP inhibited MAPK/ERK pathway by decreasing the phosphorylation of ERK1/2, resulting in reduced the Y-box binding protein 1 (YB-1) activation with blocking its nuclear translocation. Moreover, the up-regulation of P-gp expression, the activation of AKT/NF-κB and MAPK/ERK pathway induced by LPS was attenuated by GSP administration. Compared with PDTC and U1026, inhibitor of NF-κB and MAPK/ERK respectively, GSP showed the same tendency of down-regulating NF-κB and MAPK/ERK mediated YB-1 activities. Thus, GSP reverses P-gp associated MDR by inhibiting the function and expression of P-gp through down-regulation of NF-κB activity and MAPK/ERK pathway mediated YB-1 nuclear translocation, offering insight into the mechanism of reversing MDR by natural polyphenol supplement compounds. GSP could be a new potential MDR reversal agent used for combination therapy with chemotherapeutics in clinic.
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a promising candidate for the treatment of cancer, because it preferentially induces apoptosis in numerous cancer cells with little or no effect on normal cells. 5,7-Dihydroxyflavone is a dietary flavonoid commonly found in many plants. Here we show that the combined treatment with 5,7-dihydroxyflavone and TRAIL at subtoxic concentrations induced strong apoptotic response in human hepatocarcinoma HepG2 cells, acute leukemia Jurkat T cells, and cervical carcinoma HeLa cells. We further investigated the mechanisms by which 5,7-dihydroxyflavone augments TRAIL-induced apoptosis in HepG2 cells. 5,7-Dihydroxyflavone up-regulated the expression of pro-apoptotic protein Bax, attenuated the expression of anti-apoptotic proteins Bcl-2, Mcl-1, and IAPs, and reduced the phosphorylation levels of Akt and STAT3, weakening the anti-apoptotic signals thus facilitating the process of apoptosis. Moreover, 5,7-dihydroxyflavone and TRAIL were well tolerated in mice, and the combination of 5,7-dihydroxyflavone and TRAIL reduced tumor burden in vivo in a HepG2 tumor xenograft model. Interestingly, 5,7-dihydroxyflavone-mediated sensitization to TRAIL-induced cell death was not observed in normal human hepatocytes L-O2. These results suggest that the 5,7-dihydroxyflavone in combination with TRAIL might be used for cancer prevention and/or therapy.
Treatment trends of retinoblastoma (RB) have gradually evolved from eye enucleation and external radiation to local treatment. Combined treatment with an oncolytic virus and chemotherapy is currently a new method in RB treatment. To investigate the therapeutic effect of oncolytic adenovirus SG600 in combination with vincristine (VCR) on retinoblastoma in vitro, the cell viability, cell cycle effects and apoptotic activity of HXO-RB(44) cells treated with SG600, VCR or SG600 plus VCR were measured using a cell counting kit-8-based procedure and flow cytometry. Western blot analysis for Akt, p-Akt, p-p53 and p-Rb protein was performed to investigate the underlying mechanisms of combined therapy. The combination therapy exerted a synergistic antitumor effect via a type of G(2)/M and S phase arrest rather than the induction of apoptosis. The combination of VCR and SG600 further reduced Akt phosphorylation compared with cells treated with VCR alone, suggesting that SG600 could overcome chemoresistance, perhaps by down-regulating Akt in RB cells. An increase in the expression of p-p53 and decrease in p-Rb expression in HXO-RB(44) after co-treatment might be associated with cell cycle block. Western blot examination revealed that VCR might enhance SG600 replication. These results suggest that viro-chemo combination therapy is a feasible and potentially promising approach for the treatment of retinoblastoma.
To study the roles of microRNA-223 (miR-223) in regulation of cell growth, we established a miR-223 over-expression model in HeLa cells infected with miR-223 by Lentivirus pLL3.7 system. We observed in this model that miR-223 significantly suppressed the proliferation, growth rate, colony formation of HeLa cells in vitro, and in vivo tumorigenicity or tumor formation in nude mice. To investigate the mechanisms involved, we scanned and examined the potential and putative target molecules of miR-223 by informatics, quantitative PCR and Western blot, and found that insulin-like growth factor-1 receptor (IGF-1R) was the functional target of miR-223 inhibition of cell proliferation. Targeting IGF-1R by miR-223 was not only seen in HeLa cells, but also in leukemia and hepatoma cells. The downstream pathway, Akt/mTOR/p70S6K, to which the signal was mediated by IGF-1R, was inhibited as well. The relative luciferase activity of the reporter containing wild-type 3'UTR(3'untranslated region) of IGF-1R was significantly suppressed, but the mutant not. Silence of IGF-1R expression by vector-based short hairpin RNA resulted in the similar inhibition with miR-223. Contrarily, rescued IGF-1R expression in the cells that over-expressed miR-223, reversed the inhibition caused by miR-223 via introducing IGF-1R cDNA that didn't contain the 3'UTR. Meanwhile, we also noted that miR-223 targeted Rasa1, but the downstream molecules mediated by Rasa1 was neither targeted nor regulated. Therefore we believed that IGF-1R was the functional target for miR-223 suppression of cell proliferation and its downstream PI3K/Akt/mTOR/p70S6K pathway suppressed by miR-223 was by targeting IGF-1R.
BACKGROUND:
MicroRNAs (miRNAs) are frequently dysregulated in human cancers and can act as either potent oncogenes or tumor suppressor genes. In the present study, we intend to prove that the gene PTEN (phosphatase and tensin homolog deleted on chromosome ten) is a target gene of miR-205 and to investigate the suppressive effects on PTEN transcriptional activity by enhancing miR-205 expression in endometrial cancer Ishikawa cells.
METHODS:
Using Ishikawa cells as model systems, we up-regulated miR-205 expression by transient transfection with miR-205 mimics. A luciferase reporter assay, qRT-PCR and western blotting assays were used to verify whether PTEN is a direct target of miR-205. Meanwhile, the modulatory role of miR-205 in the AKT (protein kinase B) pathway was evaluated by determining the AKT phosphorylation. As a biological counterpart, we investigated cell apoptosis using flow cytometry.
RESULTS:
Our data indicate that miR-205 down-regulates the expression of PTEN through direct interaction with the putative binding site in the 3'-untranslated region (3'-UTR) of PTEN. Moreover, we documented the functional interactions of miR-205 and PTEN, which have a downstream effect on the regulation of the AKT pathway, explaining, at least in part, the inhibitory effects on Ishikawa cell apoptosis of enhancing miR-205 expression.
CONCLUSIONS:
For the first time, we demonstrate that the expression of PTEN is directly regulated by miR-205 in endometrial cancer cells and leads the inhibition of cellular apoptosis. This relationship could be targeted for new therapeutic strategies for endometrial cancer.
BACKGROUND:
The dipeptidyl peptidase-4 inhibitor sitagliptin, a new anti-diabetic medicine, is effective in treating type 2 diabetes mellitus by increasing the activation and duration of action of glucagon-like peptide-1. Since atherosclerosis is the main pathological feature of diabetic cardiovascular complications, it is important to investigate the anti-atherosclerotic effect of sitagliptin and explore the relevant mechanisms.
METHODS:
Male apolipoprotein-E-knockout mice were randomly divided into two groups and fed either high-fat diet (HFD) or HFD plus sitagliptin at a concentration of 0.3% for 16 weeks. Body weight, food intake, blood glucose, serum lipids and adhesion molecules were measured. The atherosclerotic plaque area and its histological composition were analyzed using Sudan staining and immunohistochemistry. The expression of inflammatory cytokines (monocyte chemoattractant protein (MCP)-1 and interleukin (IL)-6) and the activation of AMP-activated protein kinase (AMPK) and mitogen-activated protein kinase (MAPK) in the aortas were determined using quantitative polymerase chain reaction and western blot, respectively.
RESULTS:
Mice treated with sitagliptin developed fewer atherosclerotic plaques than the control group (7.64 ± 1.98% vs 12.91 ± 1.15%, p < 0.001), particularly in the aortic arch and abdominal aorta, where plaques were decreased 1.92- and 2.74-fold, respectively (p < 0.05 and p < 0.01). Sitagliptin significantly reduced the content of collagen fiber in plaques 1.2-fold (p < 0.05). Moreover, sitagliptin significantly reduced the expression of monocyte chemoattractant protein-1 and interleukin-6 in the aorta (p < 0.01 and p < 0.05), as well as the serum levels of soluble vascular cell adhesion molecule-1 and P-selectin (both p < 0.05). In addition, Sitagliptin induced phosphorylation of AMPK and Akt (p < 0.05 and p < 0.01), while suppressed phosphorylation of p38 and extracellular signal-regulated kinase (Erk) 1/2 (p < 0.05 and p < 0.01) in aortas.
CONCLUSIONS:
Our present study indicates that sitagliptin can reduce the area of the atherosclerotic lesion, possibly by regulating the AMPK and MAPK pathways and then reducing leukocyte -endothelial cell interaction and inflammation reactions. These actions are independent of weight loss and glucose-reducing effects.
BACKGROUND:
MT1G inactivation mediated by promoter methylation has been reported in thyroid cancer. However, the role of MT1G in thyroid carcinogenesis remains unclear. The aim of this study is to examine the biological functions and related molecular mechanisms of MT1G in thyroid cancer.
METHODS:
Methylation-specific PCR (MSP) was performed to analyze promoter methylation of MT1G and its relationship with clinicopathological characteristics of papillary thyroid cancer (PTC) patients. Conventional and real-time quantitative RT-PCR assays were used to evaluate mRNA expression. The functions of ectopic MT1G expression were determined by cell proliferation and colony formation, cell cycle and apoptosis, as well as cell migration and invasion assays.
RESULTS:
MT1G expression was frequently silenced or down-regulated in thyroid cancer cell lines, and was also significantly decreased in primary thyroid cancer tissues compared with non-malignant thyroid tissues. Promoter methylation, along with histone modification, contributes to MT1G inactivation in thyroid tumorigenesis. Moreover, our data showed that MT1G hypermethylation was significantly positively associated with lymph node metastasis in PTC patients. Importantly, restoring MT1G expression in thyroid cancer cells dramatically suppressed cell growth and invasiveness, and induced cell cycle arrest and apoptosis through inhibiting phosphorylation of Akt and Rb.
CONCLUSIONS:
We have for the first time revealed that MT1G appears to be functional tumor suppressor involved in thyroid carcinogenesis mainly through modulating the phosphatidylinositol-3-kinase (PI3K)/Akt pathway and partially through regulating the activity of Rb/E2F pathway in this study.
BACKGROUND:
The phosphoinositide 3-kinase (PI3K)/Akt pathway plays a fundamental role in cell proliferation and survival in human tumorigenesis, including gastric cancer. PIK3CA mutations and amplification are two major causes of overactivation of this pathway in human cancers. However, until this work, there was no sound investigation on the association of PIK3CA mutations and amplification with clinical outcome in gastric cancer, particularly the latter.
METHODS:
Using direct sequencing and real-time quantitative PCR, we examined PIK3CA mutations and amplification, and their association with clinicopathological characteristics and clinical outcome of gastric cancer patients.
RESULTS:
PIK3CA mutations and amplification were found in 8/113 (7.1%) and 88/131 (67%) gastric cancer patients, respectively. PIK3CA amplification was closely associated with increased phosphorylated Akt (p-Akt) level. No relationship was found between PIK3CA mutations and clinicopathological characteristics and clinical outcome in gastric cancer. PIK3CA amplification was significantly positively associated with cancer-related death. Importantly, Kaplan-Meier survival curves revealed that the patients with PIK3CA amplification had significantly shorter survival times than the patients without PIK3CA amplification.
CONCLUSIONS:
Our data showed that PIK3CA mutations were not common, but its amplification was very common in gastric cancer and may be a major mechanism in activating the PI3K/Akt pathway in gastric cancer. Importantly, Kaplan-Meier survival curves revealed that PIK3CA amplification was significantly positively associated with poor survival of gastric cancer patients. Collectively, the PI3K/Akt signaling pathway may be an effective therapeutic target in gastric cancer.
BACKGROUND:
Hepatocellular carcinoma (HCC) usually has a dismal prognosis because of its limited response to current pharmacotherapy and high metastatic rate. Sulfated oligosaccharide has been confirmed as having potent antitumor activities against solid tumors. Here, we explored the preclinical effects and molecular mechanisms of isomalto oligosaccharide sulfate (IMOS), another novel sulfated oligosaccharide, in HCC cell lines and a xenograft model.
METHODS:
The effects of IMOS on HCC proliferation, apoptosis, adhesion, migration, and invasiveness in vitro were assessed by cell counting, flow cytometry, adhesion, wound healing, and transwell assays, respectively. The roles of IMOS on HCC growth and metastasis in xenograft models were evaluated by tumor volumes and fluorescent signals. Total and phosphorylated protein levels of AKT, ERK, and JNK as well as total levels of c-MET were detected by Western blotting. IMOS-regulated genes were screened by quantitative reverse-transcription PCR (qRT-PCR) array in HCCLM3-red fluorescent protein (RFP) xenograft tissues and then confirmed by qRT-PCR in HepG2 and Hep3B cells.
RESULTS:
IMOS markedly inhibited cell proliferation and induced cell apoptosis of HCCLM3, HepG2, and Bel-7402 cells and also significantly suppressed cell adhesion, migration, and invasion of HCCLM3 in vitro. At doses of 60 and 90 mg/kg/d, IMOS displayed robust inhibitory effects on HCC growth and metastasis without obvious side effects in vivo. The levels of pERK, tERK, and pJNK as well as c-MET were significantly down-regulated after treatment with 16 mg/mL IMOS. No obvious changes were found in the levels of pAkt, tAkt, and tJNK. Ten differentially expressed genes were screened from HCCLM3-RFP xenograft tissues after treatment with IMOS at a dose of 90 mg/kg/d. Similar gene expression profiles were confirmed in HepG2 and Hep3B cells after treatment with 16 mg/mL IMOS.
CONCLUSIONS:
IMOS is a potential anti-HCC candidate through inhibition of ERK and JNK signaling independent of p53 and worth studying further in patients with HCC, especially at advanced stages.
Endothelial dysfunction is believed the early stage of development of diabetic cardiovascular complications. Sphingosine-1-phosphate (S1P) regulates various biological activities by binding to sphingosine-1-phosphate receptors (S1PRs) including S1PR1-S1PR5. In the present study, the role of S1P receptors in S1P-induced human coronary artery endothelial cells (HCAECs) dysfunction under high glucose condition was investigated and the underlying mechanism was explored. S1PR1-S1PR5 mRNA levels were detected by quantitative Real-time PCR. NO level and polymorphonuclear neutrophils (PMN)-endothelial cells adhesion were measured by nitrate reductase and myeloperoxidase colorimetric method, respectively. Protein levels of endothelial nitric oxide synthase (eNOS), vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1(ICAM-1), phosphatidylinositol 3-kinase (PI3K) and Akt were measured by Western blot analysis. S1PR2 were found the predominant S1P receptor expressed in HCAECs exposed to high glucose. NO level and eNOS activity were remarkably decreased, while PMN adhesion, VCAM-1 and ICAM-1 protein levels were increased significantly by S1P treatment in HCAECs exposed to high glucose and normal glucose. Blockage of S1PR2 with specific antagonist JTE-013 and small interfering RNA (siRNA) resulted in enhanced NO level and eNOS activity as well as decreased PMN adhesion, reduced protein levels of VCAM-1 and ICAM-1 induced by S1P. Furthermore, Phosphor-PI3K and phosphor-Akt level were markedly increased by S1PR2 blockade in S1P-treated cells exposed to high glucose, which were suppressed by PI3K inhibitor wortmannin. In conclusion, S1P/S1PR2 mediated endothelial dysfunction partly by inhibiting PI3K/Akt signaling pathway under high glucose condition. S1PR2 blockage could ameliorate endothelial dysfunction which might provide a potential therapeutic strategy for diabetic vascular complications.
Apelin is highly expressed in rat left ventricular hypertrophy Sprague Dawley rat models, and it plays a crucial role in the cardiovascular system. The aim this study was to clarify whether apelin-13 promotes hypertrophy in H9c2 rat cardiomyocytes and to investigate its underlying mechanism. The cardiomyocyte hypertrophy was observed by measuring the diameter, volume, and protein content of H9c2 cells. The activation of autophagy was evaluated by observing the morphology of autophagosomes by transmission electron microscopy, observing the subcellular localization of LC3 by light microscopy, and detecting the membrane-associated form of LC3 by western blot analysis. The phosphatidylinositol 3-kinase (PI3K) signaling pathway was identified and the proteins expression was detected using western blot analysis. The results revealed that apelin-13 increased the diameter, volume, and protein content of H9c2 cells and promoted the phosphorylation of PI3K, Akt, ERK1/2, and p70S6K. Apelin-13 activated the PI3K-Akt-ERK1/2-p70S6K pathway. PI3K inhibitor LY294002, Akt inhibitor 1701-1, ERK1/2 inhibitor PD98059 attenuated the increase of the cell diameter, volume, protein content induced by apelin-13. Apelin-13 increased the autophagosomes and up-regulated the expressions of beclin 1 and LC3-II/I both transiently and stably. The autophagy inhibitor 3MA ameliorated the increase of cell diameter, volume, and protein content that were induced by apelin-13. These results suggested that apelin-13 promotes H9c2 rat cardiomyocyte hypertrophy via PI3K-Akt-ERK1/2-p70S6K and PI3K-induced autophagy.
Integrin-linked kinase (ILK), a highly conserved intracellular protein of serine/threonine protein kinase activities, which is associated with the integrin and growth factor receptor signaling pathway, is involved in the regulation of cell proliferation, apoptosis, differentiation, migration and epithelial-mesenchymal transition (EMT). Findings of a previous study showed that ILK overexpression was strongly correlated with a more aggressive tumor phenotype, recurrence and poor survival for oral squamous cell carcinoma (OSCC) patients, as well as some EMT markers. In order to investigate the underlying mechanisms involved, a lentivirus-mediated short hairpin RNA (shRNA) was employed to downregulate ILK. The results showed that the knockdown of ILK inhibited cell growth, adhesion and invasion ability in vitro, and OSCC cells deficient of ILK were blocked in the S phase and underwent apoptosis. Additionally, ILK shRNA inhibited EMT by impairing the expression of Snail, Slug and Twist2 and enhacning E-cadherin expression. ILK shRNA suppressed the phosphorylation of downstream signaling targets Akt and GSk-3β. In addition, the knockdown of ILK inhibited tumor growth, invasion and metastasis of xenograft tumors in vivo. These results suggested that ILK is a promising therapeutic target for the treatment of OSCC.
Endothelial apoptosis triggered by oxidized low-density lipoprotein (oxLDL) can accelerate the progression of endothelial dysfunction atherosclerosis. Phosphocreatine (PCr) is a natural compound, which has been used in cardiac disease and cardiopulmonary resuscitation. However, its protective effects on atherosclerosis and its mechanism have not been clarified. In the present study, we investigated the anti-apoptotic effect of phosphocreatine in human umbilical vein endothelial cells (HUVECs) exposed to oxLDL and explored the possible mechanisms. HUVECs were pre-treated with 10-30 mM PCr and then stimulated with oxLDL. Cell morphology, cytotoxicity and apoptosis were evaluated by light microscopy, CCK assay, and flow cytometry respectively. Levels of Bax, Bcl-2, protein expression of protein kinase B (Akt), eNOS and caspase activities were assessed by Western blotting. Reactive oxygen species (ROS) and mitochondrial membrane potential (MMP) were measured with fluorescent probes. Lactate dehydrogenase (LDH), malondialdehyde (MDA), nitric oxide (NO) and superoxide dismutase (SOD) contents were determined by spectrophotometer. Our results showed that PCr dose-dependently prevented oxLDL associated HUVEC cytotoxicity and apoptotic biochemical changes such as loss of MMP, LDH and MDA leakage and loss of SOD, decrease of Bcl-2/Bax protein ratio, activation of caspase-3 and 9, and ROS generation. In addition, the antiapoptotic effect of PCr was partially inhibited by a PI3K inhibitor (LY294002) and also enhanced p-Akt/Akt protein ratio, eNOS activation and NO production. In conclusion, our data show that the inhibition of oxLDL-induced endothelial apoptosis by PCr is due, at least in part to its anti-oxidant activity and its ability to modulate the PI3K/Akt/eNOS signaling pathway.
Acetylcholinesterase (AChE) is impaired in brain of diabetic animals, which may be one of the reasons for diabetes-associated cognitive decline. However, the mechanism is still unknown. The present study was designed to investigate whether the increased expression of AChE in central neurons under high glucose (HG) condition was due to activation of mammalian target of rapamycin (mTOR) signaling. It was found that more production of reactive oxygen species, and higher levels of phospho-Akt, phospho-mTOR, phospho-p70S6K, and AChE were detected in HT-22 cells in HG group than normal glucose group after culture for 24 h, which were all attenuated by an antioxidant N-acetyl-L-cysteine. A PI3K inhibitor LY294002 significantly decreased the levels of phospho-Akt, phospho-mTOR, phospho-p70S6K, and AChE protein expression in HG-cultured HT-22 cells, and an mTOR inhibitor rapamycin markedly reduced the levels of phospho-mTOR, phospho-p70S6K, and AChE expression. Furthermore, compared with normal rats, diabetic rats showed remarkable increases in levels of AChE activity and expression, malondialdehyde, phospho-mTOR, phospho-p70S6K, and a significant decrease in total superoxide dismutase activity in both hippocampus and cerebral cortex. However, much lower levels of phospho-mTOR, phospho-p70S6K, and AChE expression occurred in both brain regions of diabetic rats treated with rapamycin when compared with untreated ones. These results indicated that mTOR signaling was activated through the activation of PI3K/Akt pathway mediated by oxidative stress in HG-cultured HT-22 cells and diabetic rat brains, which contributed to the elevated protein expression of AChE in central neurons under the condition of HG.
Radiotherapy is a widespread treatment in human solid tumors. However, therapy resistance and poor prognosis are still problems. Gambogic acid (GA), extracted from the dried yellow resin of gamboges, has an anticancer effect against various types of cancer cells. To explore the radiosensitivity of GA on esophageal cancer cell line TE13, cell viability was tested by Cell Counting Kit-8 (CCK-8) assay, colony formation assay was used to assess the effects of GA on the radiosensitivity of TE13, and flow cytometry was performed to meter the percentage of apoptosis. The protein levels of microtubule-associated protein 1 light chain 3 (LC3), caspase3, caspase8, casepase9, pAkt, and p-mammalian target of rapamycin (p-mTOR) were tested using Western blot. The distribution of LC3 was detected by immunofluorescence. Additionally, we also examined reactive oxygen species (ROS) expression by laser scanning confocal microscope (LSCM). The cells were transfected with adenovial vector to monitor the autophagy through the expression of green fluorescent protein (GFP-red fluroscent protein (RFP)-LC3. The rates of apoptotic cells in combined-treated TE13 increased significantly compared with the control groups in accordance with the results of Western blot. The clonogenic survival assay showed that GA enhances radiosensitivity with a sensitizing enhancement ratio (SER) of 1.217 and 1.436 at different concentrations. The LC3-II protein level increased in the combined group indicating the increase of autophagy incidence, and the results of GFP-RFP-LC3 experiment showed that GA may block the process of autophagic flux in TE13 cells. Moreover, we successfully demonstrated that ROS is involved in the induction of autophagy. ROS-mediated autophagy depends on the inhibition of the Akt/mTOR pathway. Taken together, GA induced radiosensitivity involves autophagy and apoptosis which are regulated by ROS hypergeneration and Akt/mTOR inhibition.
The aim of this study was to explore the intracellular mechanisms underlying the cardiovascular toxicity of air particulate matter (PM) with an aerodynamic diameter of less than 2.5 µm (PM2.5) in a human umbilical vein cell line, EA.hy926. We found that PM2.5 exposure triggered reactive oxygen species (ROS) generation, resulting in a significant decrease in cell viability. Data from Western blots showed that PM2.5 induced phosphorylation of Jun N-terminal kinase (JNK), extracellular signal regulatory kinase (ERK), p38 mitogen-activated protein kinase (MAPK) and protein kinase B (AKT), and activation of nuclear factor kappa B (NF-κB). We further observed a significant increase in expressions of intercellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1) in a time- and dose-dependent manner. Moreover, the adhesion of monocytic THP-1 cells to EA.hy926 cells was greatly enhanced in the presence of PM2.5 . However, N-acetylcysteine (NAC), a scavenger of ROS, prevented the increase of ROS generation, attenuated the phosphorylation of the above kinases, and decreased the NF-κB activation as well as the expression of ICAM-1 and VCAM-1. Furthermore, ERK inhibitor (U0126), AKT inhibitor (LY294002) and NF-κB inhibitor (BAY11-7082) significantly down-regulated PM2.5 -induced ICAM-1 and VCAM-1 expression as well as adhesion of THP-1 cells, but not JNK inhibitor (SP600125) and p38 MAPK inhibitor (SB203580), indicating that ERK/AKT/NF-κB is involved in the signaling pathway that leads to PM2.5 -induced ICAM-1 and VCAM-1 expression. These findings suggest PM2.5 -induced ROS may function as signaling molecules triggering ICAM-1 and VCAM-1 expressions through activating the ERK/AKT/NF-κB-dependent pathway, and further promoting monocyte adhesion to endothelial cells.
CPS-F, a polysaccharide derived from Cordyceps sinensis, is a potential anti-inflammatory and anti-oxidative agent. We demonstrated that CPS-F not only inhibits platelet-derived growth factor BB (PDGF-BB)-induced intracellular reactive oxygen species (ROS) generation, and up-regulation of tumor necrosis factor-α (TNF-α), TNF-α receptor 1 (TNFR1), and monocyte chemotactic protein-1 (MCP-1), but also acts synergistically in combination with MAPK/ERK inhibitor U0126 and PI3K/Akt inhibitor LY294002. Additionally, up-regulation of pro-inflammatory factors was reversed by use of a combination of CPS-F and NADPH oxidase (NOX) inhibitor diphenyleneiodonium chloride (DPI) or silencing of NOX1. Furthermore, CPS-F prevents the PDGF receptor β (PDGFRβ) promoter activity induced by PDGF-BB in transfected cells and ameliorates increased levels of TNF-α, TNFR1, and MCP-1 when PDGFRβ is silenced, thereby suggesting that CPS-F possesses a bidirectional regulatory function. Our findings suggest CPS-F may exert its therapeutic effect for the treatment of glomerulonephritis related to human mesangial cells (HMCs) through the ERK1/2/Akt pathways.
1. This study investigated the alteration of carboxylesterases in type 2 diabetes. We found that the carboxylesterase 1d (Ces1d) and carboxylesterase 1e (Ces1e) expression and the capacity of hydrolytic activity of liver and intestine decreased, whereas the Akt/mTOR/HIF-1α/ Stra13 (DEC1) signaling was activated in T2D mice. Consistently, high insulin could give rise to the same results in the high-glucose DMEM condition, which mimicked T2D, in primary mouse hepatocytes. 2. Perifosine or rapamycin almost abolished the decrease of the Ces1d and Ces1e expression and the hydrolytic activity induced by the insulin in the primary mouse hepatocytes. 3. The responsiveness of human hepatoma (HepG2) cells to high insulin in high-glucose condition was similar to that of primary mouse hepatocytes in terms of the altered expression of carboxylesterases. 4. The knockdown of HIF-1α or DEC1 with shRNA construct abrogated the decrease of the CES1 and CES2 expression induced by the insulin in high glucose condition in HepG2 cells. 5. Taken together, the decreased carboxylesterases expression and hydrolytic activity in T2D mice are through the Akt/mTOR/HIF-1α/Stra13 (DEC1) pathway.
The medical properties of baicalin have been well known for many years. However, the discovery that baicalin in the presence of metal ions is more effective than baicalin alone changed the course of drug research. The present study was designed to investigate the effect and possible mechanism of apoptosis induced by baicalin-copper in a human hepatoblastoma cancer cell line (HepG2) and in vivo. This study demonstrated that baicalin-copper suppresses the proliferation of HepG2 cells in a dose-dependent manner. Intraperitoneal injection of baicalin-copper resulted in a significant decrease in tumor growth in xenografts in nude mice. Acridine orange staining and flow cytometry analysis demonstrated that baicalin-copper induced apoptosis in HepG2 cells and caused cells to arrest in G2-M phase of the cell cycle. Furthermore, baicalin-copper treatment significantly increased the Bax/Bcl-2 ratio and p38 levels, as well as decreased the expression of caspase-3, p-PI3K, p-Akt and p-mTOR (P < 0.01). All of the evidences above indicate that baicalin-copper induces apoptosis in HepG2 cells by down-regulating the PI3K/Akt/mTOR signaling pathway.
Identification of immunosuppressants from natural sources has a proven track record in immune mediated disorders. Pseudolaric acid B is a diterpenoid isolated from the roots of Pseudolarix amabilis, possessing potent immunomodulatory effect. However, the cytotoxicity limits its future clinical application. The purpose of this study was to investigate the immunosuppressive activity of Hexahydropseudolaric acid B, a Pseudolaric acid B derivative, on T cell-mediated immune response both in vitro and in vivo, and investigated its immunomodulatory effect to develop a more ascendant immunosuppressive agent. The results showed that Hexahydropseudolaric acid B could exert more preferable immunosuppressive activity and lower cytotoxicity than Pseudolaric acid B. Hexahydropseudolaric acid B significantly inhibited T cell proliferation activated by mitogen and alloantigen without obvious cytotoxicity in vitro. Furthermore, Hexahydropseudolaric acid B could ameliorate ear swelling in a mouse model of 2,4-dinitrofluorobenzene-induced delayed-type hypersensitivity in vivo. Mechanistic study revealed that Hexahydropseudolaric acid B could enhance regulatory T cells via promoting Foxp3 expression and TGF-β level, accompanied by attenuating Akt activation, blocking p38MAPK/MK2-HSP27 signal cascades, and up-regulating PPAR-γ expression. Taken together, these results suggest that Hexahydropseudolaric acid B exerts more preferable immunosuppressive activity than its precursor Pseudolaric acid B by affecting multiple targets, which support the need for continued efforts to characterize the efficacy of HPAB as a promising and safe candidate to treat immune-related diseases.
Ubiquitin‑specific protease 22 (USP22) is a component of the transcription regulatory histone acetylation complex SAGA, which broadly regulates gene transcription and correlates with cancer progression, metastasis and prognosis. Autophagy is a cell pathway with dual functions that promotes cell survival or death. However, it is not known whether USP22 can regulate autophagy in pancreatic cancer. In the present study, we first identified that USP22 was overexpressed in a large number of pancreatic cancer patient samples, concomitant with the increased expression of LC3, a marker of autophagy. Statistical analysis revealed that the increase in USP22 and autophagy was positively correlated with poor prognosis of pancreatic cancer patients. Further investigation using a human pancreatic cancer cell (Panc‑1) identified that the overexpression of USP22 increased the processing of LC3 into the active form LC3‑II and the number of autophagosomes, thus leading to enhanced autophagy. Activation of ERK1/2 kinase rather than AKT1 by USP22 was found to be one of the mechanisms promoting LC3 processing. USP22‑induced autophagy was also found to enhance cell proliferation and resistance to starvation and chemotherapeutic drugs in Panc‑1 cells, therefore expressing an overall effect that promotes cell survival. Collectively, the present study demonstrated a new function of USP22 that induces autophagy, thus leading to the poor prognosis of pancreatic cancer.
Angiogenin (ANG), a member of RNase A superfamily, is the only angiogenic factor that possesses ribonucleolytic activity. Recent studies showed that the expression of ANG was elevated in various types of cancers. Accumulating evidence indicates that ANG plays an essential role in cancer progression by stimulating both cancer cell proliferation and tumor angiogenesis. Human ribonuclease inhibitor (RI), a cytoplasmic protein, is constructed almost entirely of leucine rich repeats (LRRs), which are present in a large family of proteins that are distinguished by their display of vast surface areas to foster protein-protein interactions. RI might be involved in unknown biological effects except inhibiting RNase A activity. The experiment demonstrated that RI also could suppress activity of angiogenin (ANG) through closely combining with it in vitro. PI3K/AKT/mTOR signaling pathway exerts a key role in cell growth, survival, proliferation, apoptosis and angiogenesis. We recently reported that up-regulating RI inhibited the growth and induced apoptosis of murine melanoma cells through repression of angiogenin and PI3K/AKT signaling pathway. However, ANG receptors have not yet been identified to date, its related signal transduction pathways are not fully clear and underlying interacting mechanisms between RI and ANG remain largely unknown. Therefore, we hypothesize that RI might combine with intracellular ANG to block its nuclear translocation and regulate PI3K/AKT/mTOR signaling pathway to inhibit biological functions of ANG. Here, we reported for the first time that ANG could interact with RI endogenously and exogenously by using co-immunoprecipitation (Co-IP) and GST pull-down. Furthermore, we observed the colocalization of ANG and RI in cells with immunofluorescence staining under laser confocal microscope. Moreover, through fluorescence resonance energy transfer (FRET) assay, we further confirmed that these two proteins have a physical interaction in living cells. Subsequently, we demonstrated that up-regulating ANG including ANG His37Ala mutant obviously decreased RI expression and activated phosphorylation of key downstream target molecules of PI3K/AKT/mTOR signaling pathway. Finally, up-regulating ANG led to the promotion of tumor angiogenesis, tumorigenesis and metastasis in vivo. Taken together, our data provided a novel mechanism of ANG in regulating PI3K/AKT/mTOR signaling pathway via RI, which suggested a new therapeutic target for cancer therapy.
Tenuifoliside A (TFSA) is a bioactive oligosaccharide ester component of Polygala tenuifolia Wild, a traditional Chinese medicine which was used to manage mental disorders effectively. The neuroprotective and anti-apoptotic effects of TFSA have been demonstrated in our previous studies. The present work was designed to study the molecular mechanism of TFSA on promoting the viability of rat glioma cells C6. We exposed C6 cells to TFSA (or combined with ERK, PI3K and TrkB inhibitors) to examine the effects of TFSA on the cell viability and the expression and phosphorylation of key proteins in the ERK and PI3K signaling pathway. TFSA increased levels of phospho-ERK and phospho-Akt, enhanced release of BDNF, which were blocked by ERK and PI3K inhibitors, respectively (U0126 and LY294002). Moreover, the TFSA caused the enhanced phosphorylation of cyclic AMP response element binding protein (CREB) at Ser133 site, the effect was revoked by U0126, LY294002 and K252a. Furthermore, when C6 cells were pretreated with K252a, a TrkB antagonist, known to significantly inhibit the activity of brain-derived neurotrophic factor (BDNF), blocked the levels of phospho-ERK, phospho-Akt and phosphor-CREB. Taking these results together, we suggested the neuroprotection of TFSA might be mediated through BDNF/TrkB-ERK/PI3K-CREB signaling pathway in C6 glioma cells.
Triple-negative breast cancer (TNBC) is an aggressive breast cancer with a generally poor prognosis. Due to lack of specific targets for its treatment, an efficient therapy is needed. G protein-coupled estrogen receptor (GPER), a novel estrogen receptor, has been reported to be expressed in TNBC tissues. In this study, we investigated the effects of blocking non-genomic signaling mediated by the estrogen/GPER pathway on cell viability and motility in the TNBC cells. GPER was strongly expressed in the TNBC cell lines MDA-MB-468 and MDA-MB-436, and the estrogen-mediated non-genomic ERK signaling activated by GPER was involved in cell viability and motility of TNBC cells. Treatment with 17β-estradiol (E2), the GPER-specific agonist G-1 and tamoxifen (TAM) led to rapid activation of p-ERK1/2, but not p-Akt. Moreover, estrogen/GPER/ERK signaling was involved in increasing cell growth, survival, and migration/invasion by upregulating expression of cyclinA, cyclinD1, Bcl-2, and c-fos associated with the cell cycle, proliferation, and apoptosis. Immunohistochemical analysis of TNBC specimens showed a significantly different staining of p-ERK1/2 between GPER-positive tissues (58/66, 87.9%) and GPER-negative tissues (13/30, 43.3%). The positivity of GPER and p-ERK1/2 displayed a strong association with large tumor size and poor clinical stage, indicating that GPER/ERK signaling might also contribute to tumor progression in TNBC patients which corresponded with in vitro experimental data. Our findings suggest that inhibition of estrogen/GPER/ERK signaling represents a novel targeted therapy in TNBC.
This study was carried out to investigate the impact of tripterygium glycosides (TGs) on ovarian function of female rats in vitro and in vivo. In vitro studies showed that TG induced cells decrease at G1 phase and inhibited cell proliferation in rat granulosa cells. In vivo, female rats were intragastrically administered with TG at the dose of 60 mg/kg/day for consecutive 50 days. TG caused a prolonged estrous cycle, and a significant reduction in ovarian index, serum E2 level, and numbers of secondary and antral follicles (p < 0.05) in these rats. A significant reduction of viable embryos was demonstrated in TG-treated female rats after mating (p < 0.01). Further, we observed observed the reduced expression level of TGF-β1 after TG treatment in vitro and in vivo. Moreover, the expression of Smad2 and AKT was also decreased after TG treatment. These results suggest that TG can impair ovarian function through Smads-mediated TGF-β1 signal pathway.
Human ribonuclease inhibitor (RI), a cytoplasmic protein, is constructed almost entirely of leucine rich repeats. RI could suppress activities of ribonuclease and angiogenin (ANG) through closely combining with them. ANG is a potent inducer of blood vessel growth and has been implicated in the establishment, growth, and metastasis of tumors. ILK/PI3K/AKT signaling pathway also plays important roles in cell growth, cell-cycle progression, tumor angiogenesis, and cell apoptosis. Our previous experiments demonstrated that RI might effectively inhibit some tumor growth and metastasis. Our recent study showed that ILK siRNA inhibited the growth and induced apoptosis in bladder cancer cells as well as increased RI expression, which suggest a correlation between RI and ILK. However, the exact molecular mechanism of RI in anti-tumor and in the cross-talk of ANG and ILK signaling pathway remains largely unknown. Here we investigated the effects of up-regulating RI on the growth and apoptosis in murine melanoma cells through angiogenin and ILK/PI3K/AKT signaling pathway. We demonstrated that up-regulating RI obviously decreased ANG expression and activity. We also discovered that RI overexpression could remarkably inhibit cell proliferation, regulate cell cycle and induce apoptosis. Furthermore, up-regulation of RI inhibited phosphorylation of ILK downstream signaling targets protein kinase B/Akt, glycogen synthase kinase 3-beta (GSK-3β), and reduced β-catenin expression in vivo and vitro. More importantly, RI significant inhibited the tumor growth and angiogenesis of tumor bearing C57BL/6 mice. In conclusion, our findings, for the first time, suggest that angiogenin and ILK signaling pathway plays a pivotal role in mediating the inhibitory effects of RI on melanoma cells growth. This study identifies that RI may be a useful molecular target for melanoma therapy.
The precise mechanism through which the two estrogen receptor subtypes, ERα and ERβ, are linked to endometrial malignant progression is not fully understood. The aim of the present study was to examine their role in endometrial carcinoma cell migration, invasion and proliferation. We also explored the correlation between the ERs and phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathways in endometrial carcinoma cells. Using Ishikawa and KLE cells as model systems, we performed transient transfection to upregulate ERα and ERβ expression; fluorescence microscopy analysis was then employed to evaluate transfection efficiencies, RT-PCR and western blot assays were used to evaluate the mRNA and protein levels. We further examined the effects on cell migration, invasion and proliferation. We showed that ERα raised the phosphorylation levels of PI3K p85α, activated the phosphorylation of AKT and mTOR in Ishikawa and KLE cells, but ERβ had no effect on PI3K p85α phosphorylation. Moreover, the overexpression of ERs enhanced cell migration, invasion and proliferation. The effect on the activation of the PI3K/AKT/mTOR transduction cascade by ERα explains, at least in part, the enhancement on cell invasion and proliferation exerted by overexpression of ERα. This crosstalk could be taken into account in developing novel therapeutic methods by targeting the ERα and PI3K/AKT/mTOR pathways in endometrial carcinoma.
Colorectal cancer (CRC) is the third most common cancer in the USA. MicroRNAs play important roles in the pathogenesis of CRC. In this study, we investigated the role of miR-30b in CRC and found that its expression was significantly lower in CRC tissues than that in normal tissues. We showed that a low expression level of miR-30b was closely related to poor differentiation, advanced TNM stage and poor prognosis of CRC. Further experiments showed that over-expression of miR-30b suppressed CRC cell proliferation in vitro and tumour growth in vivo. Specifically, miR-30b promoted G1 arrest and induced apoptosis. Moreover, KRAS, PIK3CD and BCL2 were identified as direct and functional targets of miR-30b. MiR-30b directly targeted the 3'-untranslated regions of their mRNAs and repressed their expression. This study revealed functional and mechanistic links between miRNA-30b and oncogene KRAS, PIK3CD and BCL2 in the pathogenesis of CRC. MiR-30b not only plays important roles in the regulation of cell proliferation and tumour growth in CRC, but is also a potential prognostic marker or therapeutic target for CRC. Restoration of miR-30b expression may represent a promising therapeutic approach for targeting malignant CRC.
Progressive death of retinal ganglion cells (RGCs) is a major cause of irreversible visual impairment after optic nerve injury. Clinically, there are still no effective treatments for recovering the visual function at present. The probable approaches to maintain the vision and RGCs function involve in preventing RGCs from death and/or promoting the regeneration of damaged RGCs. Previous studies have shown that mesenchymal stem cells (MSCs) take neuroprotective effects on ischemia-induced cortical and spinal cord injury, however, whether MSCs have a beneficial effect on the optical nerve injury is not clearly determined. In present study, we transplanted MSCs derived from human umbilical cord blood (hUCB-MSCs) into the vitreous cavity of adult rats and investigated the probable capacity of anti-apoptosis and pro-neuroprotective effects on RGCs. RGCs were retrogradely traced by fluorescent gold particles (FG); cellular apoptosis was investigated by caspase-3 immunohistochemistry and terminal dUTP nick end labeling (TUNEL) staining. Hematoxylin-eosin (HE) staining was used to observe the morphological changes of the retina. Growth associated protein 43 (GAP-43), an established marker for axonal regeneration, was used to visualize the regenerative process over time. Expression of P2X7 receptors (P2X7R), which are responsible for inflammatory and immune responses, was also monitored in our experiments. We found that the hUCB-MSC transplantation significantly decreased cellular apoptosis and promoted the survival of RGCs in early phase. However, this protection was transient and the RGCs could not be protected from death in the end. Consistent with apoptosis detection, P2X7R was also significantly decreased in hUCB-MSC transplanted rats in the early time but without obvious difference to the rats from control group in the end. Thus, our results imply that hUCB-MSCs take anti-apoptotic, pro-neuroregenerative and anti-inflammatory effects in the early time for acute optic nerve injury in adult rats but could not prevent RGCs from death eventually.
Integrin-linked kinase (ILK) is a multifunctional serine/threonine kinase. Accumulating evidences suggest that ILK are involved in cell-matrix interactions, cell proliferation, invasion, migration, angiogenesis and Epithelial-mesenchymal transition (EMT). However, the underlying mechanisms remain largely unknown. EMT has been postulated as a prerequisite for metastasis. The reports have demonstrated that EMT was implicated in metastasis of oral squamous cell carcinomas. Therefore, here we further postulate that ILK might participate in EMT of tongue cancer. We showed that ILK siRNA inhibited EMT with low N-cadherin, Vimentin, Snail, Slug and Twist as well as high E-cadherin expression in vivo and in vitro. We found that knockdown of ILK inhibited cell proliferation, migration and invasion as well as changed cell morphology. We also demonstrated that ILK siRNA inhibited phosphorylation of downstream signaling targets Akt and GSK3β as well as reduced expression of MMP2 and MMP9. Furthermore, we found that the tongue tumor with high metastasis capability showed higher ILK, Vimentin, Snail, Slug and Twist as well as lower E-cadherin expression in clinical specimens. Finally, ILK siRNA led to the suppression for tumorigenesis and metastasis in vivo. Our findings suggest that ILK could be a novel diagnostic and therapeutic target for tongue cancer.
Human ribonuclease inhibitor (RI) is a cytoplasmic acidic protein possibly involved in biological functions other than the inhibition of RNase A and angiogenin activities. We have previously shown that RI can inhibit growth and metastasis in some cancer cells. Epithelial-mesenchymal transition (EMT) is regarded as the beginning of invasion and metastasis and has been implicated in the metastasis of bladder cancer. We therefore postulate that RI regulates EMT of bladder cancer cells. We find that the over-expression of RI induces the up-regulation of E-cadherin, accompanied with the decreased expression of proteins associated with EMT, such as N-cadherin, Snail, Slug, vimentin and Twist and of matrix metalloprotein-2 (MMP-2), MMP-9 and Cyclin-D1, both in vitro and in vivo. The up-regulation of RI inhibits cell proliferation, migration and invasion, alters cell morphology and adhesion and leads to the rearrangement of the cytoskeleton in vitro. We also demonstrate that the up-regulation of RI can decrease the expression of integrin-linked kinase (ILK), a central component of signaling cascades controlling an array of biological processes. The over-expression of RI reduces the phosphorylation of the ILK downstream signaling targets p-Akt and p-GSK3β in T24 cells. We further find that bladder cancer with a high-metastasis capability shows higher vimentin, Snail, Slug and Twist and lower E-cadherin and RI expression in human clinical specimens. Finally, we provide evidence that the up-regulation of RI inhibits tumorigenesis and metastasis of bladder cancer in vivo. Thus, RI might play a novel role in the development of bladder cancer through regulating EMT and the ILK signaling pathway.
The multi-targeted therapy for liver cancer has been considered as a novel strategy to fight hepatocellular carcinoma. In this study, we first found that sprengerinin C, a naturally derived compound strongly suppressed tumor angiogenesis in human umbilical vein endothelial cells. A mechanism study revealed that sprengerinin C blocked vascular endothelial growth factor receptor 2-dependent phosphoinositide 3-kinase/Akt/mTOR/matrix metalloproteinase and p38 MAPK/matrix metalloproteinase pathways, two major pathways for tumor angiogenesis. Moreover, sprengerinin C inhibited vascular endothelial growth factor release, a vital event for early angiogenesis response, from hypoxic HepG-2/BEL7402 cells by suppressing hypoxia-inducible factor-1α transcriptional activity. Furthermore, sprengerinin C induced HepG-2/BEL7402 cell apoptosis by activating NADPH oxidase/reactive oxygen species-dependent caspase apoptosis pathway and suppressed HepG-2/BEL7402 cell growth through p53-mediated G2/M-phase arrest. Sprengerinin C also showed a significant anti-tumor effect in the nude mouse xenograft model of human hepatocellular carcinoma. These results provide new insights into development of potent candidate compounds for liver cancer through affecting multiple tumor progression steps of angiogenesis, apoptosis and proliferation.
Excessive βAR stimulation is an independent factor in inducing pathological cardiac hypertrophy. Here, we report miR-145 regulates both expression and localization of GATA6, thereby protecting the heart against cardiomyocyte hypertrophy induced by isoproterenol (ISO). The protective activity of miR-145 was associated with down-regulation of ANF, BNP and β-MHC expression, a decreased rate of protein synthesis, inhibited cardiomyocyte growth and the modulation of several signaling pathways including ERK1/2, JNK and Akt-GSK3β. The anti-hypertrophic effect was abrogated by exogenous over-expression of transcription factor GATA6 which was further identified as a direct target of miR-145. In addition, GSK3β antagonists, LiCl and TDZD8, restored the nuclear accumulation of GATA6, which was attenuated by miR-145 Finally, we observed a dynamic pattern of miR-145 expression in ISO-treated NRCMs and in the hearts of TAC mice. Together, our results identify miR-145 as an important regulator in cardiac hypertrophy.
The aim of this study was to investigate the protective effect of 5-HMF on human umbilical vein endothelial cells (HUVECs) injured by high glucose in vitro, and the mechanism underlying this process. Our results demonstrated that high glucose-induced oxidative stress in HUVECs was mainly mediated through activation of reactive oxygen species (ROS), Jun N-kinase 2/3 (JNK2/3) and plasma interleukin-8 (IL-8), and inactivation of phosphorylated protein kinase B (P-Akt). Treatment of HUVECs with media containing high glucose (4.5%) in the presence of 5-HMF (100, 200 and 400 μM) resulted in significant inhibition of high glucose-induced oxidative stress and expression of JNK1 and JNK2/3. Furthermore, 5-HMF rapidly inhibited high glucose-induced activation of IL-8, a downstream activator of P-Akt. Diabetes mellitus can cause a wide variety of vascular complications and high glucose can induce vascular endothelial cell apoptosis. Free radicals are formed disproportionately in diabetes by glucose oxidation. The finding of this study highlights the pharmacological application of 5-HMF for preventing cardiovascular and diabetes mellitus diseases, and provides the theoretical basis for further development of a Cornus officinalis agent for diabetes-associated vascular diseases.
Trimetazidine (TMZ) is a widely used drug exerting cardioprotective effects against ischemic heart disease through a number of mechanisms in conditions of oxidative stress. However, there are few data regarding the effects of TMZ on endothelial lineage, especially endothelial progenitor cells (EPCs). Thus, we sought to investigate whether TMZ could protect EPCs against oxidative stress injury induced by H2O2 (100 µM) and the preliminary mechanisms involved in vitro. The results showed that pretreatment of EPCs with TMZ (10 µM) protected the proliferation, adhesion, migration, and apoptosis of EPCs against H2O2, accompanied by an increase in superoxide dismutase (SOD) activity, a decrease in malonaldehyde (MDA) content, and increases in eNOS, Akt phosphorylation, and NO production. These TMZ-mediated beneficial effects on EPCs could be attenuated by pre-incubation with the Akt inhibitor triciribine. In conclusion, the present study demonstrates that TMZ ameliorated H2O2-induced impairment of biological functions in EPCs with the involvement of antioxidation and Akt/eNOS signaling pathway. These findings suggest that TMZ mediating preservation of EPCs may contribute to its cardioprotective effects on ischemic heart disease.
Angiogenesis has become an attractive target for the treatment of certain diseases such as cancer and rheumatoid arthritis. Our previous studies demonstrated that the saponin fraction from Gleditsia sinensis fruits had anti-angiogenic potential, and Gleditsiosides B (GB) was probably the main active constituent. In the present study, we assessed the effect of GB on endothelial cell migration, a crucial event in angiogenesis, and explored the underlying mechanisms. The migration of endothelial cells was assessed by transwell. The expressions of MMP-2/-9 and TIMP-1/-2 were analyzed by Western blotting, and the activities of MMP-2/-9 were detected by gelatin zymography assay. Moreover, migration-related proteins and signaling pathways, including FAK, MAPKs and PI3K/AKT, were analyzed by Western blotting. It was shown that GB, at a concentration of 10 μM without significant cytotoxicity, could effectively abrogate the migration of human umbilical vein endothelial cells (HUVECs) induced by bFGF. GB also inhibited the expression and activity of MMP-2, elevated the expression of TIMP-1, and restrained the phosphorylations of FAK, ERK, PI3K and AKT in a concentration-dependent manner. The findings suggest that GB was able to abrogate the migration of endothelial cells through down-regulating the activation of MMP-2 and FAK via preventing ERK and PI3K/AKT signaling pathways.
Endothelial apoptosis triggered by oxidized low-density lipoprotein (ox-LDL) can accelerate the progression of endothelial dysfunction in atherosclerosis. (±)7,8-Dihydroxy-3-methyl-isochromanone-4 (XJP-1) is a natural phenolic compound derived from banana peel. In the present study, we investigated the anti-apoptotic effect of XJP-1 in human umbilical vein endothelial cells (HUVECs) exposed to ox-LDL and explored underlying mechanisms. Our results showed that in the presence of ox-LDL, XJP-1 significantly attenuated ox-LDL-mediated cytotoxicity, apoptosis, caspase-3 activation, reactive oxygen species (ROS) generation, and NADPH oxidase subunit (p22phox and p47phox) expression in HUVECs. In addition, the anticytotoxic and anti-apoptotic effect of XJP-1 was partially inhibited by a PI3K inhibitor (LY294002), an Akt inhibitor (SH-6), a specific eNOS inhibitor (l-NAME) and a NADPH oxidase inhibitor (DPI). In exploring the underlying mechanisms of XJP-1 action, we found that XJP-1 eliminated ox-LDL-induced dephosphorylation of Akt and eNOS in a dose-dependent manner. However, XJP-1 alone upregulation of Akt and eNOS phosphorylation were blocked by LY294002 and SH-6. Moreover, XJP-1 increased NO production, but this effect was abolished by LY294002, SH-6 and l-NAME. The inhibition of ox-LDL-induced endothelial dysfunction by XJP-1 is due at least in part to its anti-oxidant activity and its ability to modulate the PI3K/Akt/eNOS signaling pathway.
Integrin-linked kinase (ILK) is a multifunctional serine/threonine kinase in cytoplasm. Recent studies showed that cancer patients with increased ILK expression had low survival, poor prognosis and increased metastasis. Although the causes of ILK overexpression remain to be fully elucidated, accumulating evidence suggests that its oncogenic capacity derives from its regulation of several downstream targets that provide cells with signals that promote proliferation, survival and migration. However, the mechanisms underlying tumor metastasis by ILK is still not fully understood. Epithelial–mesenchymal transition (EMT) is a critical event of cancer cells that triggers invasion and metastasis. We recently reported that knockdown of ILK inhibited the growth and induced apoptosis in human bladder cancer cells. Therefore, we postulate that ILK might involve in EMT. Here we further investigate the function of ILK with RNA interference in bladder cancer cells. Knockdown of ILK impeded an EMT with low Vimentin, Snail, Slug and Twist as well as high E-cadherin expression in vivo and vitro. In addition, we found that knockdown of ILK inhibited cell proliferation, migration and invasion as well as changed cell morphology, adhesion and rearranged cytoskeleton in vitro. We also demonstrated that ILK siRNA inhibited phosphorylation of downstream signaling targets Akt and GSK3β, increased expression of nm23-H1, as well as reduced expression of MMP-2 and MMP-9 in vivo and vitro. Furthermore, downregulation of ILK could increase expression of Ribonuclease inhibitor (RI), an important acidic cytoplasmic protein with many functions. Finally, the effects of ILK siRNA on bladder cancer cell phenotype and invasiveness translate into suppression for tumorigenesis and metastasis in vivo. Taken together, our findings highlight that ILK signaling pathway plays a novel role in the development of bladder cancer through regulating EMT. ILK could be a promising diagnostic marker and therapeutic target for bladder cancer.
It is well known that reactive oxygen species (ROS) plays a role in the pathogenesis of insulin resistance which is the hallmark of type 2 diabetes. However, it is still needed to clarify the mechanism underlying insulin resistance. Glucose oxidase (GOD) is an oxi-reductase catalyzing the conversion of glucose to glucolactone, which is further converted to glucuronic acid and H(2)O(2). The present study was designed to establish a rat model of insulin resistance using GOD and to investigate possible mechanisms. The results showed that three days administration of GOD could significantly increase fasting blood glucose, resulting in impaired glucose and insulin tolerance. Moreover, GOD disrupted insulin signaling both in rats and in hepatocytes, as evidenced by decreased phosphorylation of insulin-stimulated Akt, GSK3 and FOXO1α. Furthermore, GOD administration decreased the expression of PPARγ, alterated the phosphorylation of MAPKs, including p38, ERK and JNK, increased the expression of GRP78 and reduced the expression of PGC-1α and decreased the activities of ATPase and respiratory complexes, all of which have been reported to contribute to insulin resistance. Redox balance was evaluated by detecting the expression of antioxidant defenses and ROS generation. After the treatment with GOD, nuclear factorerythroid 2 p45-related factor 2 (Nrf2)-regulated antioxidant enzymes were damaged and ROS production increased significantly. N-acetyl-L-cysteine (NAC), a potent antioxidant, could notably inhibit these effects of GOD. Although further studies are needed to investigate the clear mechanism, these data also support the conclusion that, if not the most early event, ROS generation is the most important event that plays a central role in the pathogenesis of insulin resistance. Overall, our study established an insulin resistant animal model induced by GOD, elucidated the importance of ROS in pathogenesis of insulin resistance and provided the clue for further studies on the underlying mechanisms.
Cardiac hypertrophy is a response of the myocardium to increased workload and is characterised by an increase of myocardial mass and an accumulation of extracellular matrix (ECM). As an ECM protein, an integrin ligand, and an angiogenesis inhibitor, all of which are key players in cardiac hypertrophy, mindin is an attractive target for therapeutic intervention to treat or prevent cardiac hypertrophy and heart failure. In this study, we investigated the role of mindin in cardiac hypertrophy using littermate Mindin knockout (Mindin ( -/- )) and wild-type (WT) mice. Cardiac hypertrophy was induced by aortic banding (AB) or angiotensin II (Ang II) infusion in Mindin ( -/- ) and WT mice. The extent of cardiac hypertrophy was quantitated by echocardiography and by pathological and molecular analyses of heart samples. Mindin ( -/- ) mice were more susceptible to cardiac hypertrophy and fibrosis in response to AB or Ang II stimulation than wild type. Cardiac function was also markedly exacerbated during both systole and diastole in Mindin ( -/- ) mice in response to hypertrophic stimuli. Western blot assays further showed that the activation of AKT/glycogen synthase kinase 3β (GSK3β) signalling in response to hypertrophic stimuli was significantly increased in Mindin ( -/- ) mice. Moreover, blocking AKT/GSK3β signalling with a pharmacological AKT inhibitor reversed cardiac abnormalities in Mindin ( -/- ) mice. Our data show that mindin, as an intrinsic cardioprotective factor, prevents maladaptive remodelling and the transition to heart failure by blocking AKT/GSK3β signalling.
Puerarin is an isoflavonoid isolated from the root of the plant Pueraria lobata and has been used as a prescribed drug in China for the treatment of cardiovascular diseases in the clinical practice. Puerarin possesses potential therapeutic activities for metabolic and cardiovascular disorders. However, little is yet known about its bioprotection against endothelial dysfunction insulin resistance involved. In this study, we established insulin resistance by palmitate stimulation in the endothelium and investigated the action of puerarin on the modulation of insulin sensitivity under the insulin resistant condition. Palmitate stimulation impaired insulin-mediated vasodilation in the rat aorta and puerarin treatment effectively restored the impaired vasodilation in a concentration-dependent manner (1, 10 and 50 μM). Palmitate stimulation evoked inflammatory response in endothelial cells. Puerarin inhibited IKKβ/NF-κB activation and decreased TNF-α and IL-6 production with the downregulation of relative gene overexpression. Palmitate stimulation impaired the insulin PI3K signaling pathway and reduced insulin-mediated NO production in endothelial cells. Puerarin attenuated PA-induced phosphorylation of insulin receptor substrate-1 (IRS-1) at S307 and effectively ameliorated insulin-mediated tyrosine phosphorylation of IRS-1. The beneficial modification of serine/tyrosine phosphorylation of IRS-1 restored downstream Akt/eNOS activation, and thereby increased insulin-mediated NO production. These results suggest that puerarin inhibits inflammation and attenuates endothelial insulin resistance in an IKKβ/IRS-1-dependent manner.
Oxidative stress plays a fundamental role in the development of diabetes, which has become a great threaten for health in the whole world. In recent years, it has been found that, in addition to the effect on metabolism, insulin plays an antioxidant role. However, the effect of insulin on the whole antioxidant enzyme system, especially in vivo, is not completely understood. We note that, in vitro and in vivo, insulin administration could sequentially and transiently increase a battery of antioxidant enzymes through the activation of the key transcription factor nuclear factor erythroid 2 p45-related factor 2 (Nrf2). The sequential activation of extracellular signal-regulated kinases (ERK)-protein kinase B (Akt) pathway maybe required for insulin-induced enhancement of antioxidant defense regulated by Nrf2. Our observation leads to the hypothesis that insulin regulates the redox balance and insulin bolsters antioxidant defenses via the ERK-Akt-Nrf2 pathway.
Artemisinin (Art) is a sesquiterpene trioxane lactone from Artemisia annua L., which has been shown to affect immune responses. However, the underlying mechanism remains elusive. In this study, we examined the anti-inflammatory and immunomodulatory effects of Art in a mouse model of contact hypersensitivity (CHS), a T-cell-mediated cutaneous inflammatory reaction. The data showed that topical administration of Art could suppress CHS response and Con A-induced T cell proliferation effectively. Further experiments indicated that Art induced the generation of regulatory T cells (Tregs) and impaired the phosphorylation of AKT, associated with the up-regulation of p38 MAPK activation. Moreover, Art also exerted a strikingly inhibitory effect on IL-17 production, and diminished the level of IL-6 paralleled with an enhancement of TGF-β, which effects were coupled with a significant reduction of STAT3 activation. These data reveal that Art could effectively block CHS response in mice by inducing the generation of Tregs and suppressing the development of Th17, indicating the potential of Art to be applied as an effective therapeutic agent for treating immune-related diseases.
Previous work in our laboratory has shown that scopoletin, one of the main bioactive constituents of Erycibe obtusifolia Benth stems, exerts anti-arthritic activity in vivo partly by preventing synovial angiogenesis. The present study was performed to further investigate the anti-angiogenic potential of scopoletin, focusing on the mechanisms of action in vitro. In the aortic ring sprouting assay, scopoletin (10, 30 and 100 μM) significantly inhibited the growth of endothelial sprouts in a concentration-dependent manner. As to human umbilical vein endothelial cells (HUVECs), scopoletin could inhibit their proliferation, migration and tubule formation induced by FGF-2, especially the proliferation. It also remarkably decreased the expression of VEGF at mRNA and protein levels, and the phosphorylations of IKKα and IκB but not Akt, as well as the degradation of IκB caused by FGF-2 in HUVECs. These findings suggest that scopoletin is substantially able to attenuate FGF-2-induced angiogenesis, and it might act by directly preventing the stimulation action of FGF-2 and by indirectly decreasing the production of VEGF. Scopoletin down-regulated the VEGF expression through NF-κB rather than PI-3K/Akt signaling pathway.
We examined the effects of anti-six-transmembrane epithelial antigen of the prostate-4 (STEAP4) antibodies on glucose transport in mature adipocytes and determined the mechanism of insulin resistance in obesity. Western blotting was performed to determine STEAP4 expression, to assess translocation of insulin-sensitive glucose transporter 4 (GLUT4), and to measure phosphorylation and total protein content of insulin-signaling proteins. Confocal laser microscopy and flow cytometry were used to detect intracellular reactive oxygen species (ROS) and fluctuations in mitochondrial membrane potential (ΔΨ). ATP production was measured by using a luciferase-based luminescence assay kit. After the application of anti-STEAP4 antibodies at 0.002 mg/mL, adipocytes exhibited reduced insulin-stimulated glucose transport by attenuating the phosphorylation of IRS-1, PI3K (p85), and Akt. The antibodies also potentially increase the level of ROS and decrease cellular ATP production and ΔΨ. In conclusion, (i) STEAP4 regulates the function of IRS-1, PI3K, and Akt and decreases insulin-induced GLUT4 translocation and glucose uptake; (ii) ROS-related mitochondrial dysfunction may be related to a reduced IRS-1 correlation with the PI3K signaling pathway, leading to insulin resistance. These observations highlight the potential role of STEAP4 in glucose homeostasis and possibly in the pathophysiology of type 2 diabetes related to obesity and may provide new insights into the mechanisms of insulin resistance in obesity.
Integrin-linked kinase (ILK), an intracellular serine/threonine kinase, is implicated in cell growth and survival, cell-cycle progression, tumor angiogenesis, and cell apoptosis. Recent studies showed that the expression and activity of ILK increased significantly in many types of solid tumors. However, the exact molecular mechanism of ILK underlie tumor has not been fully ascertained. The purpose of our study was to determine whether knockdown of ILK would inhibit cell growth and induce apoptosis in bladder cancer cells using a plasmid vector based small interfering RNA (siRNA). The experiments showed that knockdown of ILK could remarkably inhibit cell proliferation and growth, regulate cell cycle and induce apoptosis of bladder cancer BIU-87 and EJ cells. We demonstrated that knockdown of ILK inhibited phosphorylation of downstream signaling targets protein kinase B/Akt, glycogen synthase kinase 3-beta (GSK-3β), and reduced expression of β-catenin in BIU-87 as well as EJ cells by Western blot and Immunofluorescence analysis. In addition, down-regulation of ILK also could increase expression of Ribonuclease inhibitor (RI), an important acidic cytoplasmic protein with many functions. BALB/C nude mice injected with the BIU-87 cells transfected ILK siRNA showed a significant inhibition of the tumor growth with lighter tumor weight, lower microvessels density and higher apoptosis rate than those in the other two control groups. In conclusion, these results suggest that ILK might be involved in the development of bladder cancer, and could be served as a novel potential therapy target for human bladder cancer. Our study may be of biological and clinical importance.
CaN induces the apoptosis in neurons, but the influence of CIPC and the intervention of pretreament with Ca(2+)-regulated factors, such as nimodipine, MK801 and cyclosporine A, on CaN expression is not clear. We also do not know whether cbl-b takes part in the induction of ischemia or induces an expression change of cbl-b in CIPC. So we will discuss the effect of CIPC, pretreatment with nimodipine, MK801 and cyclosporine A on the expression of the CaN, cbl-b and p-AKT in the hippocampus neurons. In our study, we established rat models including sham, ischemia, CIPC, nimodipine, MK801 and cyclosporine A. The neurological deficit scores were processed. The right hippocampus was removed and stained with TTC, and the volume of cerebral infarction was calculated. The apoptotic neurons were detected by TUNEL staining. The expressions of CaN, cbl-b and p-AKT at the protein level were examined by Western blotting, and the transcription of cbl-b by RT-PCR, respectively. The results showed that the neurological deficit scores, the volume of the cerebral infarction, the numbers of the apoptotic neurons, the protein expression of CaN, cbl-b and the transcription of cbl-b were the highest in the ischemia and MK801 groups, there were no difference between the two groups(P>0.05); these factors in CIPC group were all lower than those in the ischemia group(P<0.05); and much lower in the nimodipine and cyclosporine A group than those in the CIPC group (among them, the volume of the cerebral infarction in the nimodipine and cyclosporine A groups P<0.01, the expression of CaN in nimodipine group P<0.01, others were P<0.05), but no significant difference existed between the nimodipine and cyclosporine A groups(P>0.05). The expression of p-AKT was the lowest in the ischemia and MK801 groups, and there was no difference between the two groups (P>0.05), This factor was higher in CIPC group than that in the ischemia group (P<0.05); it was the highest in the nimodipine and cyclosporine A groups among these groups (the nimodipine group P<0.01, the cyclosporine A group P<0.05), no significant difference existed between the nimodipine and cyclosporine A groups (P>0.05. Continuous ischemia increases the expression of CaN, and the transcription and the protein expression of cbl-b. Cbl-b reduces the phosphorylated expression of AKT, ultimately activating apoptosis. CIPC inhibits above process and reduces the expression of CaN and cbl-b, and increases the expression of p-AKT, thereby inhibiting apoptosis in neurons. Nimodipine and cyclosporine A can reduce the expression of CaN and cbl-b, and increase the expression of p-AKT, via a moderate increase in the concentration of intracellular calcium and inhibition of the activity of CaN; MK801 counteracts the effect of CIPC.
Oleanolic acid (OA), a widely used plant-derived triterpenoid, has been shown to possess potent antiatherosclerotic effects, which may be associated with the induction of heme oxygenase-1 (HO-1). However, the underlying mechanisms involved in the effect of OA on HO-1 expression are unclear. In the current study, primary rat vascular smooth muscle cells (VSMCs) were exposed to OA and we found that it enhanced HO-1 expression in a concentration- and time-dependent manner, accompanied by increased HO-1 activity. VSMCs treated with OA exhibited activation of Akt, p38 and extracellular-signal-regulated kinase (ERK). Wortmannin (a PI3K inhibitor) and PD98059 (an ERK inhibitor) attenuated OA-induced HO-1 expression, whereas SB203580 (a p38 inhibitor) had no effect. The transcription factor NF-E2-related factor 2 (Nrf2) is a key regulator of HO-1 expression. OA treatment increased Nrf2 nuclear translocation, which was also inhibited by wortmannin and PD98059. Furthermore, transfection of VSMCs with the Nrf2 siRNA-expressing lentiviral vector decreased HO-1 expression induced by OA. Finally, pretreatment of VSMCs with OA remarkably reduced hydrogen peroxide-induced cell apoptotic death, and this effect was greatly attenuated in the presence of ZnPP (a HO-1 inhibitor), wortmannin or PD98059. Taken together, these results suggest that activation of Akt and ERK is required for OA-induced activation of Nrf2 followed by upregulation of HO-1 expression in VSMCs, which may confer an adaptive survival response in atherosclerosis.
Endothelial insulin resistance is tightly associated with diabetic cardiovascular complication, and it is well known that inflammation plays an important role in the development of insulin resistance. Luteolin, a flavonoid abundant in some medical and eatable plants, is a potent inhibitor of inflammation. It is also reported that luteolin exhibited some chemoprotection capability to the endothelial integrity. This study aims to clarify whether the anti-inflammatory potency of luteolin contributes to amelioration of insulin resistance in the endothelium. Palmitate (PA) stimulation markedly reduced insulin-mediated endothelium-dependent relaxation in rat aorta, while luteolin pretreatment effectively reversed the effects of palmitate in a concentration-dependent manner. PA stimulation also evoked inflammatory response in endothelial cells. When the cells were pretreated with luteolin, IKKβ phosphorylation were reduced, which, in turn, blocked the NF-κB activation through attenuating P65 phosphorylation. At the same time, it was also found that the gene over-expressions for TNF-α and IL-6 were also reduced by luteolin pretreatment. When endothelial cells were stimulated with PA, the insulin signaling cascades were impaired with reduced insulin-dependent production of NO. Again, pretreatment of luteolin could effectively reverse the effects of PA. Luteolin modulated the Ser/Thr phosphorylation of insulin receptor substrates-1 and restored downstream Akt/eNOS activation, resulting in increased NO production in the presence of insulin. In conclusion, these results suggested that luteolin ameliorated inflammation related endothelial insulin resistance in an IKKβ/IRS-1/Akt/eNOS-dependent pathway.
Our previous studies revealed that scopoletin, the main bioactive constituent of Erycibe obtusifolia Benth stems, exerted anti-arthritic activity in vivo partly by preventing synovial angiogenesis. Herein we further investigated the anti-angiogenic potential and related mechanisms of this coumarin compound in vivo and in vitro. On chick chorioallantoic membrane (CAM) model, scopoletin (10, 30, 100 nmol/egg) dose-dependently reduced the blood vessels that were quantified by counting the number of blood vessel branch points. In vitro, scopoletin at concentrations above 30 microM obviously inhibited the VEGF-induced tube formation, proliferation and migration of human umbilical vein endothelial cells (HUVECs). Furthermore, scopoletin was shown to block VEGF-induced autophosphorylation of VEGFR2 but not VEGFR1, and down-regulate the following activation of ERK1/2, p38 MAPK and endothelial nitric oxide synthase (eNOS) as well as the production of nitric oxide (NO) in HUVECs. In sum, our findings further support that scopoletin is a candidate of angiogenesis inhibitors, and it functions by interrupting the autophosphorylation of VEGF receptor 2 (VEGFR2) and the downstream signaling pathways.
Oxidative stress is a major cause in neurodegenerative diseases including Alzheimer's disease (AD), Parkinson's disease (PD), and cerebral ischemia. Ginsenoside Rg1, a natural product extracted from Panax ginseng C.A. Meyer, has been reported to exert notable neuroprotective activities, which partly ascribed to its antioxidative activity. However, its molecular mechanism against oxidative stress induced by exogenous hydrogen peroxide (H(2)O(2)) remained unclear. In this study, we investigated its effect on H(2)O(2)-induced cell death and explored possible signaling pathway in PC12 cells. We proved that pretreatment with Rg1 at concentrations of 0.1-10 μM remarkably reduced the cytotoxicity induced by 400 μM of H(2)O(2) in PC12 cells by MTT and Hoechst and PI double staining assay. Of note, we demonstrated the activation of NF-κB signaling pathway induced by H(2)O(2) thoroughly in PC12 cells, and Rg1 suppressed phosphorylation and nuclear translocation of NF-κB/p65, phosphorylation and degradation of inhibitor protein of κB (IκB) as well as the phosphorylation of IκB-kinase complex (IKK) by western blotting or indirect immunofluorescence assay. Besides, Rg1 also inhibited the activation of Akt and the extracellular signal-regulated kinase 1/2 (ERK1/2). Furthermore, the protection of Rg1 on H(2)O(2)-injured PC12 cells was attenuated by pretreatment with two NF-κB pathway inhibitors (JSH-23 or BOT-64). In conclusion, our results suggest that Rg1 could rescue the cell injury by H(2)O(2) via down-regulation NF-κB signaling pathway as well as Akt and ERK1/2 activation, which put new evidence on the neuroprotective mechanism of Rg1 against the oxidative stress and the regulatory role of H(2)O(2) in NF-κB pathway in PC12 cells.
ETHNOPHARMACOLOGICAL RELEVANCE:
Acupuncture is a key part of traditional Chinese medicine, shown to induce favorable neuroplasticity for injuries in the central and peripheral nervous systems. Recent studies report elongated needle therapy (ENT) with BL54 and ST28 may restore acute spinal cord injury (ASCI). However, the precise mechanism for this has not been elucidated.
AIM OF THE STUDY:
In our current study, we investigated the effects of ENT on inflammation and neuronal apoptosis induced by ASCI, and whether PI3K/Akt and MAPK/ERK signaling pathways are involved in the ENT restoration effect.
MATERIALS AND METHODS:
Rat models of moderate SCI were established in accordance with the modified Allen's method and were treated with ENT continuously for 7 days. Spontaneous activities were evaluated by the Basso Beattie and Bresnahan locomotor scale. Levels of inflammatory cytokines, such as tumor necrosis factor alpha, interleukin-6, IL-1β, and nuclear factor kappa-β, were determined by enzyme-linked immunosorbent assay. Cell apoptosis was examined by the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. The proportions of cells with positive Bcl-2 and Bax expression were determined by immunohistochemical assays, whilst the expression profiles of p-AKT and p-ERK in spinal cord tissues were evaluated by western blotting. Furthermore, the expression profiles of Cytochrome-C (Cyt-C) and caspase-3 in vivo were analyzed by reverse transcription polymerase chain reaction. The potential inhibitory effects downstream of the Akt and ERK signaling pathways were examined by administration of specific inhibitors LY294002 and PD98059 in vivo.
RESULTS:
As indicated by this study, inflammation as well as PI3K/Akt- and MAPK/ERK signaling pathway-mediated neuronal apoptosis were involved in the course of SCI in rats. The neuro-protective effect of ENT was associated with reduced Bax protein-positive neurons and increased Bcl-2 protein-positive neurons. ENT enhanced recovery of rat activities. Activation of p-Akt and p-ERK in the PI3K/Akt and MAPK/ERK signaling pathways, inhibited expression of the critical component Cyt-C. Cyt-C is required for the mitochondrial apoptosis pathway and cascade of caspase-3, which is involved in activation of neuronal apoptosis through down-regulation of Bax protein and up-regulation of Bcl-2, as determined by TUNEL. The administration of PI3K/Akt and MAPK/ERK signaling pathway specific inhibitors, LY294002 and PD98059, suppressed expression of both p-Akt and p-ERK.
CONCLUSION:
ENT with BL54 and ST28 points can promote the recovery of ASCI. And the neuro-protective effect of ENT during the restoration of SCI may be associated with the suppression of both inflammation and activation of PI3K/Akt and MAPK/ERK signaling pathways, resulting from down-regulation of Bax protein, up-regulation of Bcl-2, and inhibition of the mitochondrial apoptosis pathway.
Copyright © 2016. Published by Elsevier Ireland Ltd.
BACKGROUND:
Tao-Hong-Si-Wu decoction (THSWD) is a traditional Chinese herbal medicine that has been used for centuries in the treatment of Chinese patients with chronic liver disease. Recently, THSWD has been reported to alter vascular endothelial growth factor (VEGF) induced angiogenesis, raising the possibility that in addition to its anti-inflammatory properties; THSWD might also inhibit hepatic blood flow associated fibrosis.
AIM:
To document the effects of THSWD on hepatic necroinflammatory disease activity, fibrosis and VEGF signaling in a murine model of chronic liver disease.
METHODS:
Sixty adult mice were equally divided into six study groups. Five groups were exposed to subcutaneous carbon tetrachloride (0.1 ml/10 g BW) for six weeks. Three of the five groups were treated with different concentrations of THSWD (4.25, 8.50, 17.00 g/kg), one with 0.1mg/kg of Colchicine (positive control), and one with physiologic saline (negative control). Mice in the sixth group were not exposed to CCl4 and remained untreated (healthy controls). Liver enzymes/function tests, hyaluronic acid and laminin levels were measured in serum, and hepatic histology, VEGF, Flt-1 and kinase insert domain-containing receptor (KDR), Akt and phosphorylated Akt (pAkt) expression were documented in liver tissue at the end of treatment.
RESULTS:
Hepatic necroinflammatory disease activity and fibrosis were significantly attenuated in THSWD treated mice in a dose dependent manner. These beneficial results were similar and often exceeded those achieved with Colchicine. In addition, VEGF, Flt-1, KDR, Akt and pAkt mRNA and protein expression were reduced in TSHWD treated mice.
CONCLUSIONS:
In this animal model of chronic liver disease, THSWD decreased hepatic necroinflammatory disease and fibrosis. Inhibition of VEGF expression and downstream signaling were associated with these findings. Further studies with this and other TCMs as treatment for chronic liver disease are warranted.
Copyright © 2016 Elsevier Ireland Ltd. All rights reserved.
BACKGROUND:
Recently, extract of Ginkgo biloba leaves (GbE) have become widely known phytomedicines and have shown various pharmacological activities, including improvement of blood circulation, protection of oxidative cell damage, prevention of Alzheimer's disease, treatment of cardiovascular disease and diabetes complications. This study was designed to investigate the effects of an ethanolic GbE on renal fibrosis in diabetic nephropathy (DN) and to clarify the possible mechanism by which GbE prevents renal fibrosis.
STUDY DESIGN:
We investigated the protective effects of GbE on renal fibrosis in STZ-induced diabetic rats. Rats were randomized into six groups termed normal control, diabetes mellitus, low dose of GbE (50 mg/kg/d), intermediate dose of GbE (100 mg/kg/d), high dose of GbE (200 mg/kg/d) and rapamycin (1 mg/kg/d).
METHODS:
After 12 weeks, the rats were sacrificed and then fasting blood glucose (FBG), creatinine (Cr), blood urea nitrogen (BUN), urine protein, relative kidney weight, glycogen and collagen accumulation, and collagen IV and laminin expression were measured by different methods. The amounts of E-cadherin, α-SMA and snail, as well as the phosphorylation of Akt, mTOR and p70S6K in the renal cortex of rats, were examined by western blotting.
RESULTS:
Compared with diabetic rats, the levels of Cr, BUN, urine protein, relative kidney weight, accumulation of glycogen and collagen, and expression of collagen IV and laminin in the renal cortex were all decreased in GbE treated rats. In addition, GbE reduced the expression of E-cadherin, α-SMA, snail and the phosphorylation of Akt, mTOR and p70S6K in diabetic renal cortex.
CONCLUSION:
GbE can prevent renal fibrosis in rats with diabetic nephropathy, which is most likely to be associated with its abilities to inhibit the Akt/mTOR signaling pathway.
Copyright © 2015 Elsevier GmbH. All rights reserved.
BACKGROUND:
Considering the key role of TF in coagulation of sepsis or acute lung injury (ALI), we investigated whether berberine (BBR) could inhibit TF expression and procoagulant activity and explored its possible mechanism.
METHODS:
The effects of berberine on the expression, procoagulant activity of TF and related signal pathways induced by lipopolysaccharide (LPS) were observed in THP-1 cells.
RESULTS:
Our results showed that berberine could inhibit LPS-induced TF activity and expression, and down-regulate NF-κB, Akt and MAPK/JNK/p38/ERK pathways.
CONCLUSION:
Berberine inhibits TF expression and related pathway, which provides some new insights on its mechanism for sepsis treatment.
Copyright © 2014 Institute of Pharmacology, Polish Academy of Sciences. Published by Elsevier Urban & Partner Sp. z o.o. All rights reserved.
BACKGROUND AND AIM:
Accumulating clinical evidence suggests that hyperuricemia is strongly associated with abnormal glucose metabolism and insulin resistance. However, how high uric acid (HUA) level causes insulin resistance remains unclear. We aimed to determine the direct role of HUA in insulin resistance in vitro and in vivo in mice.
METHODS:
An acute hyperuricemia mouse model was created by potassium oxonate treatment, and the impact of HUA level on insulin resistance was investigated by glucose tolerance test, insulin tolerance test and insulin signalling, including phosphorylation of insulin receptor substrate 1 (IRS1) and Akt. HepG2 cells were exposed to HUA treatment and N-acetylcysteine (NAC), reactive oxygen species scavenger; IRS1 and Akt phosphorylation was detected by Western blot analysis after insulin treatment.
RESULTS:
Hyperuricemic mice showed impaired glucose tolerance with insulin resistance. Hyperuricemia inhibited phospho-Akt (Ser473) response to insulin and increased phosphor-IRS1 (Ser307) in liver, muscle and fat tissues. HUA induced oxidative stress, and the antioxidant NAC blocked HUA-induced IRS1 activation and Akt inhibition in HepG2 cells.
CONCLUSION:
This study supplies the first evidence of HUA directly inducing insulin resistance in vivo and in vitro. Increased uric acid level may inhibit IRS1 and Akt insulin signalling and induce insulin resistance. The reactive oxygen species pathway plays a key role in HUA-induced insulin resistance.
Copyright © 2014 Elsevier Inc. All rights reserved.
BACKGROUND:
Pancreatic cancer treatment is limited and effective drugs are needed. We investigated cucurmosin (CUS)-induced apoptosis in cystic fibrosis pancreatic adenocarcinoma cells (CFPAC-1) and a possible mechanism of action to evaluate the clinical application potential of this new Type I ribosome-inactivating protein.
METHODS:
We analyzed the growth inhibition and apoptosis of CFPAC-1 cells via methylthiazol tetrazolium assay and fluorescence-activated cell sorting. Western blot was used to analyze the protein levels of caspase 3, bcl-2, caspase 9, platelet-derived growth factor receptor (PDGFR)-β, PI3K, Akt, p-Akt, the mammalian target of rapamycin (mTOR), p-mTOR, P70S6K-α, p-P70S6K-α, 4E-BP1, p-4E-BP1 and p-Bad after CUS intervention. The mRNA expression of PDGFR-β was analyzed using reverse transcription polymerase chain reaction.
RESULTS:
CUS inhibited the proliferation of pancreatic cancer cells. The induction of apoptosis depended on the CUS dose and incubation time. The drug inhibited all of the examined proteins in the PI3K/Akt/mTOR signalling pathway and induced the active fragments of caspase 3 and caspase 9. CUS downregulated PDGFR-β expression but no significant change was observed at the mRNA level.
CONCLUSION:
CUS strongly inhibits the growth of CFPAC-1 by inducing cell apoptosis. CUS downregulated the expression of PDGFR-β at the protein level and induced the apoptosis of CFPAC-1 through the PI3K/Akt/mTOR signalling pathway.
BACKGROUND:
Cannabinoid type 2 (CB2) receptor agonists can protect myocardium against ischemia/reperfusion (I/R) injury although the underlying mechanism remains unclear. Here we report the antiapoptotic effect of CB2 receptor agonist, JWH133, during myocardial ischemia/reperfusion injury and potential underlying mechanisms.
METHODS:
Ischemia was performed by blocking left coronary artery of rat for 30 min. After ischemia for 30 min, the rat heart was reperfused for 120 min by loosing the ligation of blocking left coronary artery. JWH133 (20 mg/kg), a CB2 receptor selective agonist, or vehicles were injected intravenously 5 minutes before ischemia. Infarct size of myocardium was assessed by histological stain, myocardial apoptosis index (AI) was determined by TUNEL, and mitochondrial membrane potential (∆Ψm) was measured by flow cytometry. Western blots were performed to measure the cytochrome c release, cleaved caspase 3, cleaved caspase 9 and PI3K/Akt kinase phosphorylation.
RESULTS:
JWH133 significantly reduced the infarct size and AI of myocardium suffering I/R compared to vehicle-treated group. Further mechanistic study revealed that activation of CB2 receptor by JWH133 inhibited the loss of ΔΨm, reduction of the cleaved caspases-3 and -9, release of mitochondrial cytochrome c to the cytosol, and increase of phosphorylated Akt. These JWH133-mediated effects could be totally abrogated by PI3K inhibitor wortmanin or CB2 receptor antagonist AM630.
CONCLUSION:
Our results demonstrate that activation of CB2 receptor by JWH133 prevent apoptosis during ischemia/reperfusion through inhibition of the intrinsic mitochondria-mediated apoptotic pathway and involvement of the PI3K/Akt signal pathway.
Copyright © 2013 S. Karger AG, Basel.
BACKGROUND:
Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) presents great challenges in the treatment of non-small cell lung cancer (NSCLC) patients, while the mechanisms are still not well understood. The β-catenin signaling pathway has been found to be associated with chemoresistance and can activate the EGFR and its downstream pathways. This study aimed to investigate the role of β-catenin in acquired resistance to EGFR-TKIs in NSCLC cell lines.
METHODS:
The expression and transcriptional activity of β-catenin were measured in both the NSCLC cell line PC9 and its sub-line PC9/AB(2) which has acquired resistance to gefitinib. Knockdown and overexpression of β-catenin in the PC9/AB(2) and PC9 cells were performed. The cell survival rate and the activation of the EGFR and its downstream pathways were detected in the two cell lines after transfection.
RESULTS:
Nuclear translocation of β-catenin was increased in the PC9/AB(2) cells and the baseline expression of members of the β-catenin signaling pathway was also higher in the PC9/AB(2) cells. Knocking down the expression of β-catenin increased the sensitivity of the PC9/AB(2) cells to gefitinib by blocking the activation of the EGFR downstream pathways, while β-catenin overexpression improved PC9 cells resistance to gefitinib by enhancing the activation of the EGFR and its downstream signaling.
CONCLUSION:
β-catenin plays an important role in acquired resistance to EGFR-TKIs in NSCLC cell lines and may be a potential therapeutic target for NSCLC patients who have failed to respond to targeted therapy.
Crown Copyright © 2013. Published by Elsevier Ltd. All rights reserved.
OBJECTIVES:
Chitinase 3-like 1 (CHI3L1) is associated with poor prognosis of various human cancers. However, the clinical and prognostic significance of CHI3L1 in hepatocellular carcinoma (HCC) is largely unknown. The aim of the present study is to investigate the expression of CHI3L1 in human HCC cell lines, clinical HCC specimens and its association with expressions of phosphorylated-Akt (p-Akt), E-cadherin and prognostic significance.
METHODS:
The protein level of CHI3L1 in HCC cell lines was evaluated by western blot. The mRNA and protein levels of CHI3L1 in 19 self-paired HCC specimens were assessed by RT-PCR and western blot assays. The clinical and prognostic significance of CHI3L1 in 70 cases of HCC patients was determined by immunohistochemistry. In addition, expressions of p-Akt and E-cadherin were also assessed.
RESULTS:
The protein level of CHI3L1 paralleled with increased malignant potential of HCC cell lines (P < 0.05). The mRNA and protein levels of CHI3L1 in HCC tissues were up-regulated compared with those in adjacent peritumoral tissues and further increased in tumors with metastasis (P < 0.05). Clinicopathological analysis showed that positive CHI3L1 expression was significantly associated with larger tumor size, capsular invasion, advanced TNM stages and status of metastasis (P = 0.035, 0.003, 0.023 and 0.003, respectively). Furthermore, CHI3L1 expression was positively correlated with high level of p-Akt (r = 0.293, P = 0.014), but inversely correlated with expression of E-cadherin (r = -0.267, P = 0.026). Additionally, Kaplan-Meier survival analysis showed that HCC patients with positive CHI3L1 expression had a worse overall survival and disease-free survival compared with those with negative CHI3L1 expression (P < 0.001, respectively). Multivariate analysis identified CHI3L1 as an independent prognostic predictor for overall survival and disease-free survival of HCC patients (P = 0.044 and 0.031, respectively).
CONCLUSIONS:
CHI3L1 plays an essential role in HCC malignancies and may be served as a valuable prognostic biomarker for HCC patients.
SCOPE:
Flavonoids have well-known antioxidant, anti-inflammatory, and anti-cancer activities. Isoflavone genistein is considered a potent antioxidant agent against oxidative stress. Although several mechanisms have been proposed, a clear antioxidant mechanism of genistein is still remained to be answered.
METHODS AND RESULTS:
In this study, we focused on the concerted effects on expression of Nrf2 and phase II enzyme pathway components. Transient transfection assays, RT-PCR and immunoblot analysis were performed to study its molecular mechanisms of action. In Caco-2 cells, treatment with genistein markedly attenuated H(2)O(2) -induced peroxide formation; this amelioration was reversed by buthionine sulfoximine(GCLC inhibitor) and zinc protoporphyrin(HO-1 inhibitor). Genistein increased HO-1 and GCLC mRNA and protein expression. Genistein treatment activated the ERK1/2 and PKC signaling pathway; therefore increased Nrf2 mRNA and protein expression. The roles of the ERK1/2 and PKC signaling pathway were determined using PD98059 (ERK1/2 inhibitor) and GF109203X (PKC inhibitor) and RNA interference directed against Nrf2. Both inhibitors and siNrf2 abolished genistein-induced HO-1 and GCLC protein expression. These results suggest the involvement of ERK1/2, PKC, and Nrf2 in inducing HO-1 and GCLC by genistein.
CONCLUSION:
Our studies show that genistein up-regulated HO-1 and GCLC expression through the EKR1/2 and PKC /Nrf2 pathways during oxidative stress.
© 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
BACKGROUND:
Recent researches document that an oncogenic role of USP22 activation may contribute to progression and predict the prognosis. We have reported that USP22 mediates cell survival and proliferation by promoting the expression of BMI-1 and upregulation of activated AKT pathway in colon cancer cells. However, little is known about its mechanisms in non-small-cell lung cancer (NSCLC). Here the authors investigated the significance of activation of USP22 and potential targets BMI-1, PTEN and phospho-AKT (p-AKT) in NSCLC.
METHODS:
Expression levels of USP22, BMI-1, PTEN and p-AKT in samples from 114 patients with NSCLC were evaluated immunohistochemically using the tissue microarray method. Clinical significance was analyzed by multivariate Cox regression analysis, Kaplan-Meier curves and the log-rank test.
RESULTS:
Immunohistochemically, USP22, BMI-1, p-AKT and PTEN were positive in 66.66%, 78.07%, 71.92% and 43.85% of NSCLC samples, respectively. Statistical correlation analysis showed USP22 to be significantly correlated with BMI-1 (r=0.315, P=0.001), p-AKT (r=0.271, P=0.003), and PTEN (r=-0.384, P<0.0001). NSCLCs with positive expression of USP22, BMI-1, p-AKT, and negative expression of PTEN were significantly correlated to tumor size (P=0.0240), differentiation (P=0.0457), pT classification (P=0.0077), pN classification (P=0.0064), and AJCC stage (P=0.0363) and poor overall survival (P<0.001). Multivariate Cox proportional hazards model analysis showed that the combined 4 markers was the independent prognostic indicator of overall survival (P<0.001; HR, 5.974; 95% CI, 3.307-10.791).
CONCLUSIONS:
The simultaneous targeting of USP22, and its downstream signal transduction molecules seem highly informative in stratification of the cancer into subgroups with distinct likelihood of therapy failure, which contribute to make decision process regarding the individualized therapy selection and optimization.
Copyright © 2012 Elsevier Ireland Ltd. All rights reserved.
PURPOSE:
This study was to investigate the clinicopathologic significance and potential role of HOXB7 in the development and progression of colorectal cancer (CRC).
EXPERIMENTAL DESIGN:
The relationship between HOXB7 expression and clinical characteristics of CRC was analyzed in 224 paraffin-embedded archived CRC specimens by immunohistochemistry (IHC). The effects of HOXB7 on cell growth and proliferation, as well as on tumorigenesis, were examined both in vitro and in vivo, using MTT assay, colony formation assay, cell cycle analysis, soft agar assay, and tumorigenesis in nude mice. Western blotting and real-time reverse transcriptase-PCR were performed to examine the impact of HOXB7 on the PI3K/Akt and MAPK signaling pathways.
RESULTS:
HOXB7 protein level was significantly correlated with advanced Dukes stage (P < 0.001), T stage (P = 0.012), distant metastasis (P = 0.042), higher proliferation index (P = 0.007) and poor survival of patients (P = 0.005). Enforced expression of HOXB7 in CRC cell lines significantly enhanced cell growth, proliferation and tumorigenesis. Conversely, knockdown of HOXB7 caused an inhibition of cell growth, proliferation, and tumorigenesis. We also showed that HOXB7 accelerated G(0)-G(1) to S-phase transition concomitantly with upregulation of cyclin D1 and downregulation of p27Kip1. On the contrary, knockdown of HOXB7 caused G(1)-S-phase arrest, downregulation of cyclin D1 and upregulation of p27Kip1. Enforced expression of HOXB7 could enhance PI3K/AKT and MAPK pathway activity.
CONCLUSION:
Our findings suggest that HOXB7 protein, as a valuable marker of CRC prognosis, plays an important role in the development and progression of human CRC.
©2011 AACR.
AIM OF THE STUDY:
Modified Si-Miao-San (mSMS) has showed anti-inflammatory potency and has been used in the clinic to treat metabolic disorders such as obesity and diabetes, but whether its anti-inflammatory activity contributes to improving insulin resistance remains to be determined. This study aims to investigate the mechanistic relationship between its anti-inflammatory activity and modulation of insulin sensitivity in free fatty acid-stimulated HepG2 cells.
MATERIALS AND METHODS:
HepG2 cells were stimulated with palmitate (PA) and the effect of mSMS on insulin mediated-glycogen synthesis and triglyceride secretion was observed. The inhibition of mSMS on gene expression of proinflammatory cytokine tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and inhibitor of NF-κB kinase-β (IKKβ) activation was investigated. In addition, the effects of mSMS on insulin signaling transduction along insulin receptor substrates-1 (IRS-1)/Akt pathway were also evaluated. Furthermore, the effect of mSMS on glucose intolerance induced by conditioned-medium derived from activated macrophages was also assessed in normoglycemic mice.
RESULTS:
Treatment of hepatocytes with PA reduced insulin sensitivity and mSMS effectively increased insulin-mediated glycogen synthesis and restored insulin inhibition of triglyceride secretion. mSMS suppressed IKKβ activation and down-regulated TNF-α and IL-6 gene over-expression, demonstrating its anti-inflammatory activity in hepatocytes. PA-evoked inflammation impaired insulin signaling cascades and mSMS improved insulin signaling transduction by modification of Ser/Thr phosphorylation of IRS-1 and downstream Akt (T308), thereby improved insulin sensitivity in hepatocytes. mSMS also improved glucose intolerance induced by inflammatory cytokines in normoglycemic mice, which further demonstrated its modulation toward insulin sensitivity in vivo.
CONCLUSIONS:
The results suggest that mSMS inhibited inflammatory response and improved insulin sensitivity in hepatocytes via an IKKβ/IRS-1/Akt-dependent pathway.
Crown Copyright © 2011. Published by Elsevier Ireland Ltd. All rights reserved.
BACKGROUND:
Previous studies showed that connective tissue growth factor (CTGF)-induced proliferation of lung fibroblasts and production of chemokines in mesangial cells could be inhibited by lipoxin A(4) (LXA(4)). It is speculated that LXA(4) could modulate the CTGF-induced epithelial to mesenchymal transition.
METHODS:
The expressions of alpha-smooth muscle actin (alpha-SMA), E-cadherin, integrin-linked kinase (ILK), extracellular signal-regulated kinase 1/2 (ERK1/2), phosphatidylinositol 3-kinase (PI3-K), Akt and Smad signaling were assessed by Western blot and/or real-time RT-PCR, and activation of Ras or ILK by activity assay, expressions of alpha-SMA and zonula occludens-1 by immunofluorescence assay in proximal tubular epithelial cells (HK-2).
RESULTS:
Pretreatment of HK-2 cells with LXA(4) inhibited the morphological fibroblast-like changes and alpha-SMA expression induced by CTGF but not by transforming growth factor-beta(1) (TGF-beta(1)). The expressions of E-cadherin and zonula occludens-1 reduced by CTGF but not by TGF-beta(1) were increased by LXA(4). LXA(4) inhibited the expression and activity of ILK and activation of Ras, ERK1/2, PI3-K and Akt in HK-2 cells stimulated by CTGF. LXA(4) did not affect TGF-beta(1)-induced expression of ILK, Smad-2/3 phosphorylation and Smad-2's binding to Smad-4 and subsequent nuclear translocation.
CONCLUSION:
LXA(4) inhibits the tubular epithelial to mesenchymal transition, initiated by CTGF but not by TGF-beta(1), via downregulation of ILK, Ras/MEK/ERK1/2 and PI3-K/Akt-dependent signal pathway stimulated by CTGF.
2010 S. Karger AG, Basel.
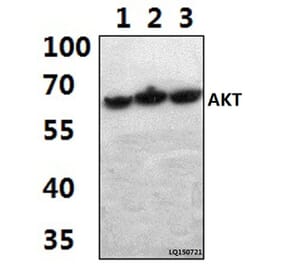
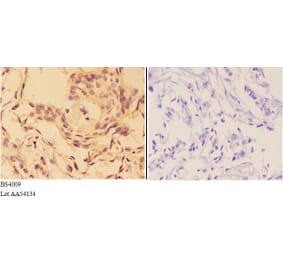
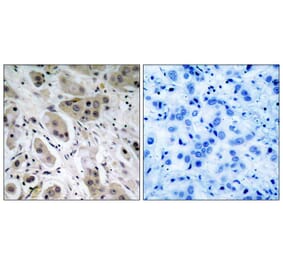
![Western Blot - Anti-AKT (phospho Ser473) Antibody [RM251] (A121248) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121248_1.png?profile=product_alternative)

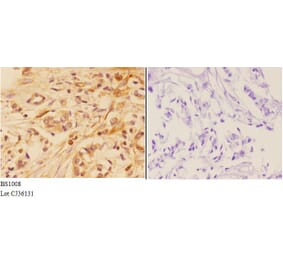
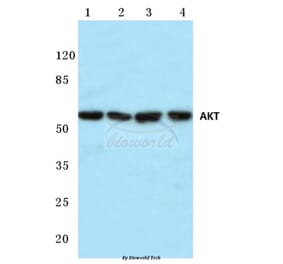

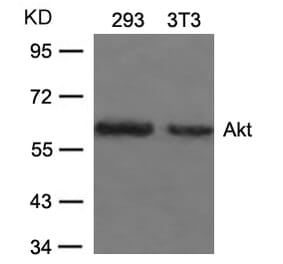


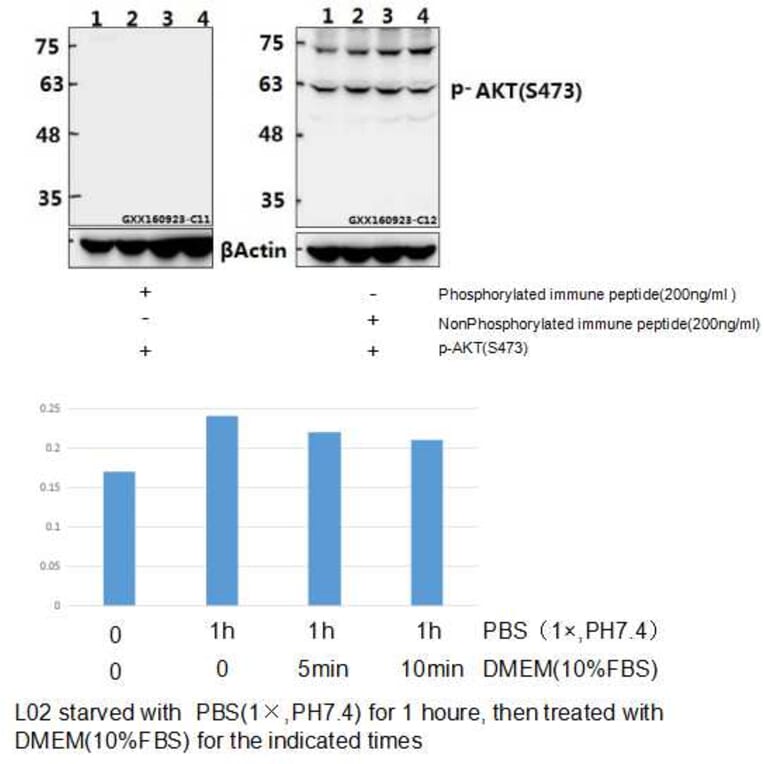



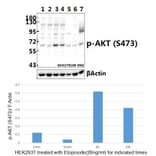




































![Immunohistochemistry - Anti-AKT Antibody [RM316] (A121372) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121394_1.png?profile=product_alternative)