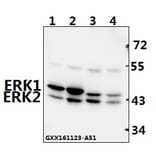
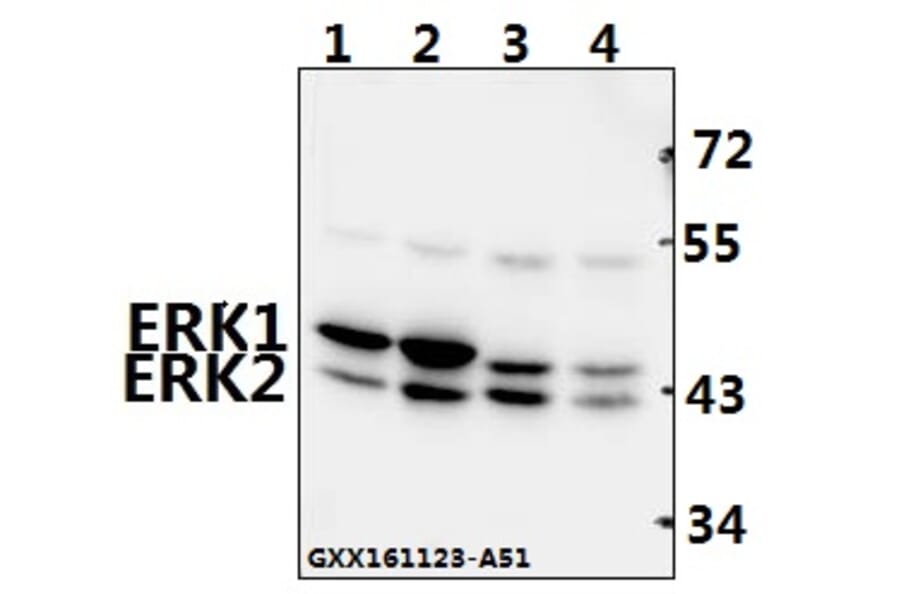
Unconjugated
Accumulating evidence indicates that heat shock protein (HSP) 60 is strongly associated with the pathology of atherosclerosis (AS). However, the precise mechanisms by which HSP60 promotes atherosclerosis remain unclear. In the present study, we found that HSP60 mRNA and protein expression levels in the thoracic aorta are enhanced not only in a mouse model of AS but also in high-fat diet (HFD) mice. HSP60 expression and secretion was activated by platelet-derived growth factor-BB (PDGF-BB) and interleukin (IL)-8 in both human umbilical vein endothelial cells (HUVECs) and vascular smooth muscle cells (VSMCs). HSP60 was found to induce VSMC migration, and exposure to HSP60 activated ERK MAPK signaling. U0126, an inhibitor of ERK, reduced VSMC migration. The HSP60-stimulated VSMCs were found to express TLR4 mRNA but not TLR2 mRNA. Knockdown of TLR4 by siRNA reduced HSP60-induced VSMC migration and HSP60-induced ERK activation. Finally, HSP60 induced IL-8 secretion in VSMCs. Together these results suggest that HSP60 is involved in the stimulation of VSMC migration, via TLR4 and ERK MAPK activation. Meanwhile, activation of HSP60 is one of the most powerful methods of sending a 'danger signal' to the immune system to generate IL-8, which assists in the management of an infection or disease.
The protein tyrosine phosphatase 1B (PTP1B), a non-transmembrane protein tyrosine phosphatase, has been implicated in gastric pathogenesis. Several lines of recent evidences have shown that PTP1B is highly amplified in breast and prostate cancers. The aim of this study was to investigate PTP1B amplification in gastric cancer and its association with poor prognosis of gastric cancer patients, and further determine the role of PTP1B in gastric tumorigenesis. Our data demonstrated that PTP1B was significantly up-regulated in gastric cancer tissues as compared with matched normal gastric tissues by using quantitative RT-PCR (qRT-PCR) assay. In addition, copy number analysis showed that PTP1B was amplified in 68/131 (51.9%) gastric cancer cases, whereas no amplification was found in the control subjects. Notably, PTP1B amplification was positively associated with its protein expression, and was significantly related to poor survival of gastric cancer patients. Knocking down PTP1B expression in gastric cancer cells significantly inhibited cell proliferation, colony formation, migration and invasion, and induced cell cycle arrested and apoptosis. Mechanically, PTP1B promotes gastric cancer cell proliferation, survival and invasiveness through modulating Src-related signaling pathways, such as Src/Ras/MAPK and Src/phosphatidylinositol-3-kinase (PI3K)/Akt pathways. Collectively, our data demonstrated frequent overexpression and amplification PTP1B in gastric cancer, and further determined the oncogenic role of PTP1B in gastric carcinogenesis. Importantly, PTP1B amplification predicts poor survival of gastric cancer patients.
Emerging evidence indicate that mesenchymal stem cells (MSCs) affect tumor progression by reshaping the tumor microenvironment. Neutrophils are essential component of the tumor microenvironment and are critically involved in cancer progression. Whether the phenotype and function of neutrophils is influenced by MSCs is not well understood. Herein, we investigated the interaction between neutrophils and gastric cancer-derived MSCs (GC-MSCs) and explored the biological role of this interaction. We found that GC-MSCs induced the chemotaxis of neutrophils and protected them from spontaneous apoptosis. Neutrophils were activated by the conditioned medium from GC-MSCs with increased expression of IL-8, TNFα, CCL2, and oncostatin M (OSM). GC-MSCs-primed neutrophils augmented the migration of gastric cancer cells in a cell contact-dependent manner but had minimal effect on gastric cancer cell proliferation. In addition, GC-MSCs-primed neutrophils prompted endothelial cells to form tube-like structure in vitro. We demonstrated that GC-MSCs stimulated the activation of STAT3 and ERK1/2 pathways in neutrophils, which was essential for the functions of activated neutrophils. We further revealed that GC-MSCs-derived IL-6 was responsible for the protection and activation of neutrophils. In turn, GC-MSCs-primed neutrophils induced the differentiation of normal MSCs into cancer-associated fibroblasts (CAFs). Collectively, our results suggest that GC-MSCs regulate the chemotaxis, survival, activation, and function of neutrophils in gastric cancer via an IL-6-STAT3-ERK1/2 signaling cascade. The reciprocal interaction between GC-MSCs and neutrophils presents a novel mechanism for the role of MSCs in remodeling cancer niche and provides a potential target for gastric cancer therapy.
It has been documented in in vitro studies that zinc oxide nanoparticles (ZnO NPs) are capable of inducing oxidative stress, which plays a crucial role in ZnO NP-mediated apoptosis. However, the underlying molecular mechanism of apoptosis in neurocytes induced by ZnO NP exposure was not fully elucidated. In this study, we investigated the potential mechanisms of apoptosis provoked by ZnO NPs in cultured primary astrocytes by exploring the molecular signaling pathways triggered after ZnO NP exposure. ZnO NP exposure was found to reduce cell viability in MTT assays, increase lactate dehydrogenase (LDH) release, stimulate intracellular reactive oxygen species (ROS) generation, and elicit caspase-3 activation in a dose- and time-dependent manner. Apoptosis occurred after ZnO NP exposure as evidenced by nuclear condensation and poly(ADP-ribose) polymerase-1 (PARP) cleavage. A decrease in mitochondrial membrane potential (MMP) with a concomitant increase in the expression of Bax/Bcl-2 ratio suggested that the mitochondria also mediated the pathway involved in ZnO NP-induced apoptosis. In addition, exposure of the cultured cells to ZnO NPs led to phosphorylation of c-Jun N-terminal kinase (JNK), extracellular signal-related kinase (ERK), and p38 mitogen-activated protein kinase (p38 MAPK). Moreover, JNK inhibitor (SP600125) significantly reduced ZnO NP-induced cleaved PARP and cleaved caspase-3 expression, but not ERK inhibitor (U0126) or p38 MAPK inhibitor (SB203580), indicating that JNK signaling pathway is involved in ZnO NP-induced apoptosis in primary astrocytes.
Cancer cell invasion, one of the crucial events in local growth and metastatic spread of tumors, possess a broad spectrum of mechanisms, especially altered expression of matrix metalloproteinases. LFG-500 is a novel synthesized flavonoid with strong anti-cancer activity, whose exact molecular mechanism remains incompletely understood. This current study was designed to examine the effects of LFG-500 on tumor metastasis using in vitro and in vivo assays. LFG-500 could inhibit adhesion, migration and invasion of MDA-MB-231 human breast carcinoma cells. Meanwhile, it reduced the activities and expression of MMP-2 and MMP-9 via suppressing the transcriptional activation of NF-κB rather than AP-1 or STAT3. Moreover, LFG-500 repressed TNF-α induced cell invasion through inhibiting NF-κB and subsequent MMP-9 activity. Further elucidation of the mechanism revealed that PI3K/AKT but not MAPK signaling pathway was involved in the inhibitory effect of LFG-500 on NF-κB activation. LFG-500 could also suppress lung metastasis of B16F10 murine melanoma cells in vivo. Taken together, these results demonstrated that LFG-500 could block cancer cell invasion via down-regulation of PI3K/AKT/NF-κB signaling pathway, which provides new evidence for the anti-cancer activity of LFG-500.
Cancer-associated fibroblasts (CAFs) are crucial co-mediators of breast cancer progression. Estrogen is the predominant driving force in the cyclic regulation of the mammary extracellular matrix, thus potentially affecting the tumor-associated stroma. Recently, a third estrogen receptor, estrogen (G-protein-coupled) receptor (GPER), has been reported to be expressed in breast CAFs. In this study, GPER was detected by immunohistochemical analysis in stromal fibroblasts of 41.8% (59/141) of the primary breast cancer samples. GPER expression in CAFs isolated from primary breast cancer tissues was confirmed by immunostaining and RT-PCR analyses. Tamoxifen (TAM) in addition to 17β-estradiol (E₂) and the GPER agonist G1 activated GPER, resulting in transient increases in cell index, intracellular calcium, and ERK1/2 phosphorylation. Furthermore, TAM, E₂, and G1 promoted CAF proliferation and cell-cycle progression, both of which were blocked by GPER interference, the selective GPER antagonist G15, the epidermal growth factor receptor (EGFR) inhibitor AG1478, and the ERK1/2 inhibitor U0126. Importantly, TAM as well as G1 increased E₂ production in breast CAFs via GPER/EGFR/ERK signaling when the substrate of E₂, testosterone, was added to the medium. GPER-induced aromatase upregulation was probably responsible for this phenomenon, as TAM- and G1-induced CYP19A1 gene expression was reduced by GPER knockdown and G15, AG1478, and U0126 administration. Accordingly, GPER-mediated CAF-dependent estrogenic effects on the tumor-associated stroma are conceivable, and CAF is likely to contribute to breast cancer progression, especially TAM resistance, via a positive feedback loop involving GPER/EGFR/ERK signaling and E₂ production.
MicroRNAs (miRNAs) have been suggested to play a vital role in regulate tumor progression and invasion. However, the expression of miR-339-5p in colorectal cancer and its effects are not known. Here, we report that miR-339-5p is a tumor suppressor by regulating expression of PRL-1. In this study, we showed that downregulated miR-339-5p levels in colorectal cancer tissues and highly invasive CRC cell lines. Furthermore, enhancing the expression of miR-339-5p inhibited CRC cell growth, migration and invasion in vitro and suppressed tumor growth in vivo. We then screened and identified a novel miR-339-5p target, phosphatases of regenerating liver-1 1 (PRL-1), and it was further confirmed by luciferase assay. Overexpression of miR-339-5p would also reduce the expression of PRL-1 mRNA and protein. The reduced PRL-1 expression was associated with low expression of phosphorylated-extracellular signal-regulated kinase1/2 (p-ERK1/2). Conversely, reduction of miR-339-5p by inhibitors in cells stimulated these phenotypes. In conclusion, our results demonstrate that miR-339-5p functions as a tumor suppressor and plays a role in inhibiting growth and metastasis of CRC cells through targeting PRL-1 and regulating p-ERK1/2 .These findings suggest that miR-339-5p may be useful as a new potential therapeutic target for CRC.
Extracellular signal-regulated kinase (ERK) belongs to the mitogen-activated protein kinases (MAPK) superfamily. Aberrant upregulation and activation of ERK cascades may often lead to tumor cell development. However, how ERK is involved in tumor progression is yet to be defined. In current study, we described that ERK undergoes S-nitrosylation by nitric oxide (NO). ERK S-nitrosylation inhibits its phosphorylation and triggers apoptotic program as verified by massive apoptosis in fluorescence staining. The proapoptotic effect of NO induced S-nitrosylation is reversed by NO scavenger Haemoglobin (HB). Furthermore, an S-nitrosylation dead ERK mutant C183A also demolishes the proapoptotic potential of NO and favors cell survival. Therefore, Cys(183) might be a potential S-nitrosylation site in ERK. In addition, S-nitrosylation is a general phenomenon that regulates ERK activity. These findings identify a novel link between NO-mediated S-nitrosylation and ERK regulation, which provide critical insights into the control of apoptosis and tumor development.
Excessive βAR stimulation is an independent factor in inducing pathological cardiac hypertrophy. Here, we report miR-145 regulates both expression and localization of GATA6, thereby protecting the heart against cardiomyocyte hypertrophy induced by isoproterenol (ISO). The protective activity of miR-145 was associated with down-regulation of ANF, BNP and β-MHC expression, a decreased rate of protein synthesis, inhibited cardiomyocyte growth and the modulation of several signaling pathways including ERK1/2, JNK and Akt-GSK3β. The anti-hypertrophic effect was abrogated by exogenous over-expression of transcription factor GATA6 which was further identified as a direct target of miR-145. In addition, GSK3β antagonists, LiCl and TDZD8, restored the nuclear accumulation of GATA6, which was attenuated by miR-145 Finally, we observed a dynamic pattern of miR-145 expression in ISO-treated NRCMs and in the hearts of TAC mice. Together, our results identify miR-145 as an important regulator in cardiac hypertrophy.
Holothurian glycosaminoglycan (hGAG) is a high-molecular-weight form of fucosylated chondroitin sulfate and has an antithrombotic effect. Our previous studies demonstrated that hGAG efficiently inhibited tumor cell metastasis. The interplays between thrombosis and tumor progression may have a major impact on hematogenous metastasis. In this study, we demonstrated that the mouse melanoma B16F10 cells treated with hGAG displayed a significant reduction of metastasis and coagulation capacity in vitro and in vivo. Mechanistic studies revealed that hGAG treatment in B16F10 cells remarkably inhibited the formation of fibrin through attenuating the generation of activated Factor Xa (FXa), without affecting the expression of urokinase (uPA) and plasminogen activator inhibitor 1 (PAI-1) that involved in fibrinolysis. Moreover, hGAG treatment downregulated the transcription and protein expression of tissue factor (TF). Promoter deletions, site mutations and functional studies identified that the nuclear transcription factor NF-κB binding region is responsible for hGAG-induced inhibition of TF expression. While the hGAG treatment of B16F10 cells was unable to inhibit NF-κB expression and phosphorylation, hGAG significantly prevented nuclear translocation of NF-κB from the cytosol, a potential mechanism underlying the transcriptional suppression of TF. Moreover, hGAG markedly suppressed the activation of p38MAPK and ERK1/2 signaling pathways, the central regulators for the expression of metastasis-related matrix metalloproteinases (MMPs). Consequently, hGAG exerts a dual function in the inhibition of metastasis and coagulation activity in mouse melanoma B16F10 cells. Our studies suggest hGAG to be a promising therapeutic agent for metastatic cancer treatment.
Hydrogen sulfide (H(2)S) has been proposed as a novel neuromodulator and neuroprotective agent. Cobalt chloride (CoCl(2)) is a well-known hypoxia mimetic agent. We have demonstrated that H(2)S protects against CoCl(2)-induced injuries in PC12 cells. However, whether the members of mitogen-activated protein kinases (MAPK), in particular, extracellular signal-regulated kinase1/2(ERK1/2) and p38MAPK are involved in the neuroprotection of H(2)S against chemical hypoxia-induced injuries of PC12 cells is not understood. We observed that CoCl(2) induced expression of transcriptional factor hypoxia-inducible factor-1 alpha (HIF-1α), decreased cystathionine-β synthase (CBS, a synthase of H(2)S) expression, and increased generation of reactive oxygen species (ROS), leading to injuries of the cells, evidenced by decrease in cell viability, dissipation of mitochondrial membrane potential (MMP) , caspase-3 activation and apoptosis, which were attenuated by pretreatment with NaHS (a donor of H(2)S) or N-acetyl-L cystein (NAC), a ROS scavenger. CoCl(2) rapidly activated ERK1/2, p38MAPK and C-Jun N-terminal kinase (JNK). Inhibition of ERK1/2 or p38MAPK or JNK with kinase inhibitors (U0126 or SB203580 or SP600125, respectively) or genetic silencing of ERK1/2 or p38MAPK by RNAi (Si-ERK1/2 or Si-p38MAPK) significantly prevented CoCl(2)-induced injuries. Pretreatment with NaHS or NAC inhibited not only CoCl(2)-induced ROS production, but also phosphorylation of ERK1/2 and p38MAPK. Thus, we demonstrated that a concurrent activation of ERK1/2, p38MAPK and JNK participates in CoCl(2)-induced injuries and that H(2)S protects PC12 cells against chemical hypoxia-induced injuries by inhibition of ROS-activated ERK1/2 and p38MAPK pathways. Our results suggest that inhibitors of ERK1/2, p38MAPK and JNK or antioxidants may be useful for preventing and treating hypoxia-induced neuronal injury.
AIM:
Plasminogen activator inhibitor-1 (PAI-1) is involved in the progression of pulmonary fibrosis. The present study was undertaken to examine the effects on pulmonary fibrosis of silencing PAI-1 expression with small interfering RNA (siRNA) and to assess the possible underlying mechanisms.
METHODS:
Male Wistar rats were subjected to intratracheal injection of bleomycin (BLM, 5 mg/kg, 0.2 mL) to induce pulmonary fibrosis. Histopathological changes of lung tissue were examined with HE or Masson's trichrome staining. The expression levels of α-smooth muscle actin (α-SMA), collagen type-I and type-III, caspase-3, as well as p-ERK1/2 and PI3K/Akt in the lung tissue were evaluated using imunohistochemistry and Western blot analyses. The fibroblasts isolated from BLM-induced fibrotic lung tissue were cultured and transfected with pcDNA-PAI-1 or PAI-1siRNA. The expression level of PAI-1 in the fibroblasts was measured using real time RT-PCR and Western blot analysis. The fibroblast proliferation was evaluated using MTT assay.
RESULTS:
Intratracheal injection of PAI-1-siRNA (7.5 nmoL/0.2 mL) significantly alleviated alveolitis and collagen deposition, reduced the expression of PAI-1, α-SMA, collagen type-I and collagen type-III, and increased the expression of caspase-3 in BLM-induced fibrotic lung tissue. In consistence with the in vivo results, the proliferation of the cultured fibroblasts from BLM-induced fibrotic lung tissue was inhibited by transfection with PAI-1-siRNA, and accelerated by overexpression of PAI-1 by transfection with pcDNA-PAI-1. The expression of caspase-3 was increased as a result of PAI-1 siRNA transfection, and decreased after transfection with pcDNA-PAI-1. In addition, the levels of p-ERK1/2 and PI3K/Akt in the fibrogenic lung tissue were reduced after treatment with PAI-1siRNA.
CONCLUSION:
The data demonstrate that PAI-1 siRNA inhibits alveolitis and pulmonary fibrosis in BLM-treated rats via inhibiting the proliferation and promoting the apoptosis of fibroblasts. Suppression ERK and AKT signalling pathways might have at least partly contributed to this process. Targeting PAI-1 is a promising therapeutic strategy for pulmonary fibrosis.
BACKGROUND:
Hepatocellular carcinoma (HCC) usually has a dismal prognosis because of its limited response to current pharmacotherapy and high metastatic rate. Sulfated oligosaccharide has been confirmed as having potent antitumor activities against solid tumors. Here, we explored the preclinical effects and molecular mechanisms of isomalto oligosaccharide sulfate (IMOS), another novel sulfated oligosaccharide, in HCC cell lines and a xenograft model.
METHODS:
The effects of IMOS on HCC proliferation, apoptosis, adhesion, migration, and invasiveness in vitro were assessed by cell counting, flow cytometry, adhesion, wound healing, and transwell assays, respectively. The roles of IMOS on HCC growth and metastasis in xenograft models were evaluated by tumor volumes and fluorescent signals. Total and phosphorylated protein levels of AKT, ERK, and JNK as well as total levels of c-MET were detected by Western blotting. IMOS-regulated genes were screened by quantitative reverse-transcription PCR (qRT-PCR) array in HCCLM3-red fluorescent protein (RFP) xenograft tissues and then confirmed by qRT-PCR in HepG2 and Hep3B cells.
RESULTS:
IMOS markedly inhibited cell proliferation and induced cell apoptosis of HCCLM3, HepG2, and Bel-7402 cells and also significantly suppressed cell adhesion, migration, and invasion of HCCLM3 in vitro. At doses of 60 and 90 mg/kg/d, IMOS displayed robust inhibitory effects on HCC growth and metastasis without obvious side effects in vivo. The levels of pERK, tERK, and pJNK as well as c-MET were significantly down-regulated after treatment with 16 mg/mL IMOS. No obvious changes were found in the levels of pAkt, tAkt, and tJNK. Ten differentially expressed genes were screened from HCCLM3-RFP xenograft tissues after treatment with IMOS at a dose of 90 mg/kg/d. Similar gene expression profiles were confirmed in HepG2 and Hep3B cells after treatment with 16 mg/mL IMOS.
CONCLUSIONS:
IMOS is a potential anti-HCC candidate through inhibition of ERK and JNK signaling independent of p53 and worth studying further in patients with HCC, especially at advanced stages.
β-cryptoxanthin (CX), a major carotenoid pigment, can inhibit inflammatory gene expression in mice with nonalcoholic steatohepatitis. In the present study, we examined the anti-inflammatory effects of CX on lipopolysaccharide (LPS)-induced inflammation in mouse primary Sertoli cells and the possible molecular mechanisms behind its effects. The results showed that CX significantly inhibited LPS-induced decreases in cell viability and in the percentage of apoptotic cells. Moreover, CX inhibited the LPS-induced up-regulation of tumor necrosis factor α (TNF-α), interleukin-10 (IL-10), interleukin-6 (IL-6) and interleukin-1β (IL-1β) in Sertoli cells. In addition, CX significantly limited the LPS-induced down-regulation of AR, HSF2, CREB, FSHR, INHBB and ABP in Sertoli cells. Western blot analysis showed that CX significantly suppressed NF-κB (p65) activation as well as MAPK phosphorylation. All the results suggested that CX suppressed inflammation, possibly associated with the NF-κB activation and MAPK of phosphorylation. Thus, CX may possess therapeutic potential against inflammation-related diseases.
Flavonoids are structurally similar to steroid hormones, particularly estrogens, and therefore have been studied for their potential effects on hormone-dependent cancers. Baicalein is the primary flavonoid derived from the root of Scutellaria baicalensis Georgi. In the present study, we investigated the effects of baicalein on 17β-estradiol (E2)-induced migration, adhesion and invasion of MCF-7 and SK-BR-3 breast cancer cells. The results demonstrated that baicalein suppressed E2-stimulated wound-healing migration and cell‑Matrigel adhesion, and ameliorated E2-promoted invasion across a Matrigel-coated Transwell membrane. Furthermore, baicalein interfered with E2-induced novel G protein-coupled estrogen receptor (GPR30)-related signaling, including a decrease in tyrosine phosphorylation of epidermal growth factor receptor (EGFR) as well as phosphorylation of extracellular signal-regulated kinase (ERK) and serine/threonine kinase Akt, without affecting GPR30 expression. The results also showed that baicalein suppressed the expression of GPR30 target genes, cysteine-rich 61 (CYR61) and connective tissue growth factor (CTGF) induced by E2. Furthermore, baicalein prevented GPR30-related signaling activation and upregulation of CYR61 and CTGF mRNA levels induced by G1, a specific GPR 30 agonist. The results suggest that baicalein inhibits E2-induced migration, adhesion and invasion through interfering with GPR30 signaling pathway activation, which indicates that it may act as a therapeutic candidate for the treatment of GPR30-positive breast cancer metastasis.
Previous studies have demonstrated that protein tyrosine phosphatase 1B (PTP1B) can promote tumor progression in breast cancer, colon cancer and prostate cancer. Additionally, PTP1B acts as a tumor suppressor in other cancers, such as esophageal cancer and lymphoma. These findings suggest that PTP1B functions as a double-facet molecule in tumors, and the role of PTP1B in non-small cell lung cancer (NSCLC) is unknown. The present study demonstrates that the expression of PTP1B in NSCLC tissue is significantly higher than its expression in benign lung disease and is associated with the stage and overall survival (OS) of NSCLC patients. In vitro studies have demonstrated that PTP1B promotes the proliferation and metastasis of NSCLC cells by reducing the expression of p-src (Tyr527), which activates src and ERK1/2. This study provides the first exploration of the role of PTP1B in the proliferation and metastasis of NSCLC and subsequently elucidates the role of PTP1B in cancer. Our study uncovered that PTP1B can promote NSCLC proliferation and metastasis by activating src and subsequently ERK1/2 and provides a theoretical basis for future applications of PTP1B inhibitors in the treatment of NSCLC.
The crude powder of the fruit of Arctium lappa L. (ALF) has previously been reported to attenuate experimental colitis in mice. But, its main effective ingredient and underlying mechanisms remain to be identified. In this study, ALF was extracted with ethanol, and then successively fractionated into petroleum ether, ethyl acetate, n-butanol and water fraction. Experimental colitis was induced by dextran sulfate sodium (DSS) in mice. Among the four fractions of ALF, the ethyl acetate fraction showed the most significant inhibition of DSS-induced colitis in mice. The comparative studies of arctigenin and arctiin (the two main ingredients of ethyl acetate fraction) indicated that arctigenin rather than arctiin could reduce the loss of body weight, disease activity index and histological damage in the colon. Arctigenin markedly recovered the loss of intestinal epithelial cells (E-cadherin-positive cells) and decreased the infiltration of neutrophils (MPO-positive cells) and macrophages (CD68-positive cells). Arctigenin could down-regulate the expressions of TNF-α, IL-6, MIP-2, MCP-1, MAdCAM-1, ICAM-1 and VCAM-1 at both protein and mRNA levels in colonic tissues. Also, it markedly decreased the MDA level, but increased SOD activity and the GSH level. Of note, the efficacy of arctigenin was comparable or even superior to that of the positive control mesalazine. Moreover, it significantly suppressed the phosphorylation of MAPKs and the activation of NF-κB, including phosphorylation of IκBα and p65, p65 translocation and DNA binding activity. In conclusion, arctigenin but not arctiin is the main active ingredient of ALF for attenuating colitis via down-regulating the activation of MAPK and NF-κB pathways.
Autophagy is a tightly-regulated catabolic pathway involving degradation of cellular proteins, cytoplasm and organelles. Recent evidence suggests that autophagy plays a potential role in cell death as a tumor suppressor and that its induction especially in combination with apoptosis could be beneficial. It remains unclear if all cancer cells behave the same mechanism when autophagy is induced. Although mammalian target of rapamycin (mTOR) is well known as a negative regulator of autophagy, the relationship between signal transducer and activator of transcription 3 (STAT3) and autophagy has not yet been investigated. Oroxylin A, a natural mono-flavonoid extracted from Scutellariae radix, is a promising therapeutic agent for treating multiple cancers. Here we investigated the mechanism underlying the effect of oroxylin A on malignant glioma cells. We showed that oroxylin A inhibited the proliferation of malignant glioma cells by inducing autophagy in a dose- and time-dependent manner. Oroxylin A treatment inhibits the AKT and ERK activation and the downstream phosphorylation level of mTOR and STAT3. In addition, oroxylin A treatment decreases the expression of Notch-1 and myeloid cell leukemia-1 (Mcl-1) but upregulates Beclin 1, the key autophagy-related protein. 3-MA (autophagy inhibitor) or knockdown of Beclin 1 partially can rescue cells from oroxylin A-induced autophagic cell death. In contrast, knockdown of STAT3 aggravates oroxylin A-induced autophagic cell death. Our data reveal an important role of autophagy in enhancing cell death induced by oroxylin A and conclude that oroxylin A exerts anti-malignant glioma proficiency by inducing autophagy via the ERK/AKT-mTOR-STAT3-Notch signaling cascade.
Tenuifoliside A (TFSA) is a bioactive oligosaccharide ester component of Polygala tenuifolia Wild, a traditional Chinese medicine which was used to manage mental disorders effectively. The neuroprotective and anti-apoptotic effects of TFSA have been demonstrated in our previous studies. The present work was designed to study the molecular mechanism of TFSA on promoting the viability of rat glioma cells C6. We exposed C6 cells to TFSA (or combined with ERK, PI3K and TrkB inhibitors) to examine the effects of TFSA on the cell viability and the expression and phosphorylation of key proteins in the ERK and PI3K signaling pathway. TFSA increased levels of phospho-ERK and phospho-Akt, enhanced release of BDNF, which were blocked by ERK and PI3K inhibitors, respectively (U0126 and LY294002). Moreover, the TFSA caused the enhanced phosphorylation of cyclic AMP response element binding protein (CREB) at Ser133 site, the effect was revoked by U0126, LY294002 and K252a. Furthermore, when C6 cells were pretreated with K252a, a TrkB antagonist, known to significantly inhibit the activity of brain-derived neurotrophic factor (BDNF), blocked the levels of phospho-ERK, phospho-Akt and phosphor-CREB. Taking these results together, we suggested the neuroprotection of TFSA might be mediated through BDNF/TrkB-ERK/PI3K-CREB signaling pathway in C6 glioma cells.
The mechanism of blue light-induced retinal ganglion cell (RGC) injury is poorly understood. In this study, we established a patented light-emitting diode-based system to study the effects of long-term blue light exposure under culture conditions on RGC-5 cells. Long-term blue light exposure significantly reduced cell viability in a time-dependent manner and induced apoptosis and necrosis in RGC-5 cells. Long-term blue light exposure marked an increase in the expression of Bax and active Caspase-3 (p17), which was accompanied by Bcl-2 down-regulation, and displayed features of the mitochondria-dependent apoptosis pathway. Blue light exposure also increased the generation of reactive oxygen species (ROS), and was a strong inducer of ROS-sensitive protein nuclear factor erythroid 2-related factor 2 (Nrf2) and heme oxygenase-1 (HO-1) expression. Moreover, blue light exposure constitutively activated p38 mitogen-activated protein kinases and c-Jun NH2-terminal kinase (JNK), as well as induced the phosphorylation of extracellular signal-regulated kinase in the early phase, in blue light-exposed RGC-5 cells. The protein expression of c-jun and c-fos was further enhanced after RGC-5 cells were exposed to blue light. Taken together, these findings indicated that blue light induced RGC-5 cell line death in dependence upon exposure duration. The potential mechanisms for this phenomenon might be via activated mitochondria-dependent apoptosis, increased ROS production and protein expressions of Nrf2 and HO-1, and activated JNK/p38 MAPK signaling pathways.
Angiogenesis is a process that forms new blood capillaries from existing vessels, which is of great physiological and pathological significance. Although recent studies provide evidence that cuprous oxide nanoparticles (CO-NPs) may have biomedical potential, the mechanisms of CO-NPs in angiogenesis have not been investigated to date. We have studied the anti-angiogenic properties of CO-NPs on primary human umbilical vein endothelial cells (HUVECs). We found that CO-NPs were able to induce cell morphology changes and suppress cell proliferation, migration and tube formation in vitro and in vivo dose dependently. Furthermore, CO-NPs could induce cell apoptosis both at the early and late apoptotic stage and induce cell cycle arrest at S phase in a dose dependent manner. As signalling via the vascular endothelial growth factor receptor-2 (VEGFR2) is critical for angiogenic responses, we further explored the expression of VEGFR2 after the treatment of CO-NPs. They were found to inhibit VEGFR2 expression dose and time dependently both at the protein and mRNA level while had no effect on VEGF and VEGFR1 expression. Together, we report for the first time that CO-NPs can act as an anti-angiogenic agent by suppressing VEGFR2 expression, which may be a potential nanomedicine for angiogenesis therapy.
Hydrolyzed fish collagen (HFC) has recently attracted considerable attention because of its outstanding bioactivity. However, few studies have been performed to determine the biological effects of HFC on bone marrow mesenchymal stem cells (BMSCs), which are often used in regenerative medicine. In this study, the molecular weight, amino acid composition, and contact angle of HFC were measured. The influence of HFC on cell viability and the multidirectional differentiation of BMSCs into osteogenic, endothelial, adipogenic, chondrogenic, and neural lineages were also assessed. Furthermore, the mechanism by which HFC promotes osteogenesis was investigated at the protein level. The molecular weight of HFC ranged from 700 to 1300 Da, the contact angle of HFC was approximately 26°, and HFC was found to be composed of various amino acids, including glycine, proline, and hydroxyproline. At a concentration of 0.2 mg/mL, HFC promoted cell viability, and significantly up-regulated the expression of osteogenic markers (RUNX2, ALP, OPN, and OCN), as well as endothelial markers (CD31, VE-cadherin, and VEGFR2). Western blot results indicated that treatment of BMSCs with 0.2 mg/mL HFC could activate the MAPK/ERK signaling pathway and then increase the protein level of RUNX2, while treatment with PD98059, a specific inhibitor of ERK1/2, could significantly inhibit the expression of P-ERK and RUNX2. Interestingly, real-time PCR demonstrated that HFC inhibited the expression of adipogenic markers (LPL and ADFP) and chondrogenic markers (aggrecan and COLII), whereas it had no effect on neural differentiation markers (MAP2 and β3-tubulin). In summary, this study suggests that without the use of any additional inducing reagent, HFC has the potential to actively promote osteogenic and endothelial differentiation because of its high hydrophilicity and the optimal extracellular microenvironment supplied by its amino acids. This research also revealed that HFC inhibited adipogenic and chondrogenic differentiation, but it had no influence on the neural differentiation of rat bone marrow mesenchymal stem cells (rBMSCs).
Multiple studies have indicated that selective cyclooxygenase-2 (COX-2) inhibitors possess clinically chemopreventive and preclinically anticancer activities. Their long-term use, however, may be limited by the cardiovascular toxicity. This study tried to investigate whether an apple oligogalactan (AOG) could enhance the growth inhibitory effect of celecoxib on colorectal cancer. Caco-2 and HT-29 cell lines were exposed to different concentrations of AOG (0-1 g/L), celecoxib (0-25 μmol/L), and their combination. COX-2 levels were assessed by reverse transcription PCR and Western blot. COX-2 activity was evaluated by measuring prostaglandin E2 concentration. A colitis-associated colorectal cancer (CACC) mouse model was used to determine the effect of the combination in vivo. AOG (0.1-0.5 g/L) could potentiate the inhibitory effect of physiologic doses of celecoxib (5 μmol/L) on cell growth and decrease COX-2 expressions both at RNA and protein levels. In vivo, the combination (2.5% AOG plus 0.04% celecoxib, w/w) prevented against CACC in mice effectively. Our data indicate that AOG could potentiate the growth inhibitory effect of celecoxib on colorectal cancer both in vitro and in vivo through influencing the expression and function of COX-2 and phosphorylation of MAPKs, which suggests a new possible combinatorial strategy in colorectal cancer therapy.
Urocortin (UCN1) is a member of corticotrophin-releasing factor (CRF) family, which has been proven to participate in inflammation. Previous work showed that dihydrotestosterone (DHT) could promote the inflammatory process. Little is known about the effect of DHT on UCN1 expression. The aim of our study is to investigate the effects and underlying mechanisms of DHT on endothelial UCN1 expression in the absence and presence of induced inflammation. Therefore, we tested the alterations of endothelial UCN1 expression treated with DHT in the presence or absence of lipopolysaccharide (LPS). Our data showed that DHT alone decreased UCN1 levels, which were attenuated in the presence of the androgen receptor (AR) antagonist flutamide. Conversely, in the presence of LPS, DHT augmented the LPS-induced increase in UCN1 expression, which was, interestingly, not affected by flutamide. When cells were treated with DHT alone, AR was upregulated and translocated into the nuclei, which might repress UCN1 expression via a potential androgen-responsive element found in human CRF family promoter. In the presence of LPS, DHT did not influence AR expression and location while it increased toll-like receptor 4 expression and activation, which was not altered by flutamide. DHT enhanced LPS-induced p38MAPK, ERK1/2, and nuclear factor κB pathway activation, which may contribute to the elevated expression of UCN1. These data suggest that DHT differentially influences UCN1 levels under normal and inflammatory conditions in human umbilical vein endothelial cells, which involves AR-dependent and -independent mechanisms respectively.
Hyperuricaemia is a disorder of purine metabolism, and is strongly associated with insulin resistance and abnormal glucose metabolism. As the producer of insulin, pancreatic β cells might be affected by elevated serum uric acid levels and contribute to the disregulated glucose metabolism. In this study, we investigated the effect of high uric acid on rat pancreatic β cell function. Under high uric acid condition, proliferation of pancreatic β cells was inhibited, production of reactive oxygen species increased, and glucose stimulated insulin secretion was also compromised. Further examination on signal transduction pathways revealed that uric acid-induced ROS is involved in the activation of adenosine monophosphate-activated protein kinase (AMPK), and extracellular signal-regulated kinase (ERK). Pharmacological inhibition of ERK activation rescued β cells from growth inhibition. More importantly, activation of ERK induced by uric acid is significantly diminished by AMPK inhibitor, indicating ERK as a downstream target of AMPK in response to high uric acid condition. We also investigated the transportation channel for uric acid into pancreatic β cells. While major urate transporter URAT1 is not expressed in β cells, organic anion transporter (OAT) inhibitor successfully blocked the activation of ERK by uric acid. Our data indicate that high uric acid levels induce oxidative damage and inhibit growth of rat pancreatic β cells by activating the AMPK and ERK signal pathways. Hyperuricemia may contribute to abnormal glucose metabolism by causing oxidative damage and function inhibition of pancreatic β cells.
An herb-derived phenolic compound, 4-hydroxybenzyl alcohol (4-HBA), exhibits beneficial effects in cerebral ischemic injury. However, the molecular mechanisms underlying this observation remain unclear. Here we used an in vitro ischemic model of oxygen-glucose deprivation followed by reperfusion (OGD/R) and an in vivo ischemic model of middle cerebral artery occlusion to investigate the relevant neuroprotective mechanisms. We demonstrated that 4-HBA reduced the neuronal injury, LDH release, and up-regulation of 8-hydroxydeoxyguanosine (8-OHdG) induced by OGD/R. Furthermore, 4-HBA reduced the cerebral infarct size and improved the behavioral parameters after cerebral ischemia. These neuroprotective effects may be conferred by the 4-HBA mediated upregulation of the transcription factor nuclear factor E2-related factor 2 (Nrf2), peroxiredoxin 6 (Prdx6) and protein disulfide isomerase (PDI) by the use of 4-HBA. Interestingly, LY294002, a phosphatidylinositol 3-kinase (PI3K) inhibitor, blocked the increase in phosphorylation of Akt and abolished the neuroprotection associated with 4-HBA. Our results suggested that 4-HBA protects neurons against cerebral ischemic injury, and this neuroprotection may occur through upregulation of Nrf2, Prdx6, and PDI expression via the PI3K/Akt pathway.
Pulmonary fibrosis is a serious and irreversible lung injury with obscure etiologic mechanisms and no effective treatment to date. This study explored a crucial link between oxidative stress and pulmonary fibrogenesis, focusing on nuclear factor erythroid 2-related factor 2 (Nrf2), a core transcription factor in antioxidative regulation systems. Treatment of C57 BL/6 mice with bleomycin increased fibroblast viability and collagen production and significantly downregulated Nrf2. In addition, prominent oxidative stress was indicated by changes in superoxide dismutase, catalase activity, and glutathione and thiobarbituric acid-reactive substance levels. In a cell-based model, bleomycin suppressed Nrf2 activation via extracellular signal-related kinase phosphorylation, enhancing intracellular reactive oxygen species in lung fibroblasts and stimulating abnormal cell proliferation and collagen secretion. To confirm this novel mechanism of bleomycin-induced fibrogenesis, we attempted to upregulate Nrf2 and related antioxidant proteins in bleomycin-treated fibroblasts using a putative Nrf2 activator, caffeic acid phenethyl ester, and the results showed that bleomycin-induced fibroblast proliferation and collagen content were attenuated through improved redox balance. Collectively, these results disclose a potential regulatory mechanism in pulmonary fibrosis that will aid the development of new therapies.
It has been well documented in in vitro studies that ambient airborne particulate matter (PM) with an aerodynamic diameter less than 2.5 μm (PM(2.5)) is capable of inducing oxidative stress, which plays a key role in PM(2.5)-mediated cytotoxicity. Although nuclear factor erythroid-2-related factor 2 (Nrf2) has been shown to regulate the intracellular defense mechanisms against oxidative stress, a potential of the Nrf2-mediated cellular defense against oxidative stress induced by PM(2.5) remains to be determined. This study was aimed to explore the potential signaling pathway of Nrf2-mediated defense mechanisms against PM(2.5)-induced oxidative stress in human type II alveolar epithelial A549 cells. We exposed A549 cells to PM(2.5) particles collected from Beijing at a concentration of 16 μg/cm(2). We observed that PM(2.5) triggered an increase of intracellular reactive oxygen species (ROS) in a time-dependent manner during a period of 2 h exposure. We also found that Nrf2 overexpression suppressed and Nrf2 knockdown increased PM(2.5)-induced ROS generation. Using Western blot and confocal microscopy, we found that PM(2.5) exposure triggered significant translocation of Nrf2 into nucleus, resulting in AKT phosphorylation and significant transcription of ARE-driven phases II enzyme genes, such as NAD(P)H:quinone oxidoreductase (NQO-1), heme oxygenase-1 (HO-1), and glutamate-cysteine ligase catalytic subunit (GCLC) in A549 cells. Evaluation of signaling pathways showed that a phosphatidylinositol 3-kinase (PI3K) inhibitor (LY294002), but not an ERK 1/2 inhibitor (PD98059) or a p38 MAPK (SB203580), significantly down-regulated PM(2.5)-induced Nrf2 nuclear translocation and HO-1 mRNA expression, indicating PI3K/AKT is involved in the signaling pathway leads to the PM(2.5)-induced nuclear translocation of Nrf2 and subsequent Nrf2-mediated HO-1 transcription. Taken together, our results suggest that PM(2.5)-induced ROS may function as signaling molecules to activate Nrf2-mediated defenses, such as HO-1 expression, against oxidative stress induced by PM(2.5) through the PI3K/AKT signaling pathway.
Formyl peptide receptor 1 (FPR1) plays an important role in the rapid progression of glioblastoma and has been considered as a molecular target for the treatment. Previously, we have shown that oligomer proanthocyanidins (F2, degree of polymerization 2-15), isolated from grape seeds, inhibited FPR1-mediated chemotaxis of U-87 glioblastoma cells. In the present study, we investigated the capacity of F2 to interact with FPR1. The cross attenuation of chemotaxis revealed that F2 shared FPR1 with formyl-methionyl-leucyl-phenylalanine (fMLF), which is a prototype agonist of FPR1. F2 was chemotactic for U-87 cells, and the chemotactic response was abolished when FPR1 gene was silenced or FPR1 was competitively occupied. We further show that F2 specifically blocked the binding of fluorescent agonist to FPR1. Interestingly, F2 exhibited the characteristic of a partial agonist for FPR1, as shown by its capacity to activate FPR1-mediated PI3K-PKC-MAPK pathways. Meanwhile, F2 also attenuated fMLF-triggered MAPK activation, suggesting that F2 could antagonize the effect of an agonist. Furthermore, F2 abolished the invasion of U-87 cells induced by fMLF. Thus, we have identified F2 as a novel, partial agonist for FPR1, which may be useful for glioblastoma therapy.
Incidence of hepatocellular carcinoma (HCC) is dramatically increasing and is the third cause of cancer death worldwide. One key approach to control HCC is chemoprevention by naturally occurring agents. This study aims at investigating the antitumor effect of oleanolic acid (OA) and the molecular mechanisms. BALB/c mice were injected subcutaneously with HepG2 cells to establish transplanted tumors. Apoptosis and cell cycle arrest-related markers and signaling cascades were determined by western blot, immunofluorescence, reverse transcriptase-polymerase chain reaction and flow cytometric analysis. OA exhibited inhibitory effect on HCC through induction of apoptosis and cell cycle arrest both in transplanted tumors and in HepG2 cells. OA induced apoptosis through mitochondrial pathway, evidenced by inhibition of Akt/mammalian target of rapamycin pathway, mitochondrial dysfunction, transient increase of adenosine triphosphate, increase of Bax/Bcl-2 ratio, increased release of cytochrome c and activation of caspase/poly (ADP-ribose) polymerase. Activation of mitochondrial apoptotic pathway may be due to reactive oxygen species generated by mitochondrial fatty acid oxidation, resulted from enhancement of lipolysis regulated by cyclic adenosine 3',5'-monophosphate response element-binding protein-hormone-sensitive lipase/peroxisome proliferator-activated receptor γ signaling. OA induced G2/M cell cycle arrest through p21-mediated downregulation of cyclin B1/cdc2. Cyclooxygenase-2 (COX-2) and p53 were involved in OA-exerted effect, and extracellular signal-regulated kinase-p53 signaling played a central role in OA-activated cascades responsible for apoptosis and cell cycle arrest. OA demonstrated significant antitumor activities in HCC in vivo and in vitro models. These data provide new insights into the mechanisms underlying the antitumor effect of OA.
Abnormal proliferation and migration of vascular smooth muscle cells (VSMC) plays an important role in vascular diseases. The Rho-associated protein kinase (ROCK) signaling pathway is now well recognized for its role in VSMC migration and proliferation. Recently, a number of studies revealed that different isoforms of ROCK have distinct functions in VSMCs. We have reported that ROCK1, rather than ROCK2, induces platelet-derived growth factor (PDGF)-BB-stimulated migration of VSMCs. In the current study, we aimed to investigate the roles of ROCK1/2 in PDGF-induced rat aorta VSMC proliferation by manipulating ROCK gene expression. The results revealed that knock-down of both ROCK1 and ROCK2 by siRNA technology decreased PDGF-BB-generated VSMC proliferation by inhibiting the expression of proliferating cell nuclear antigen (PCNA) and cyclin D1. In addition, up-regulation of ROCK1 expression through transfection, further increased the proliferation of VSMCs induced by PDGF-BB. The ERK inhibitor U0126 reduced the proliferation and expression of PCNA and cyclinD1, and ROCK1 and ROCK2 siRNA decreased the level of ERK in the nucleus. These results demonstrated that ROCK1 and ROCK2 could promote VSMC proliferation through ERK nuclear translocation, regulating the expression of PCNA and cyclin D1 protein.
HERV-K (human endogenous retrovirus type K) type 1-encoded Np9 is a tumor-specific biomarker, but its oncogenic role and targets in human leukemia remain elusive. We first identified Np9 as a potent viral oncogene in human leukemia. Silencing of Np9 inhibited the growth of myeloid and lymphoblastic leukemic cells, whereas expression of Np9 significantly promoted the growth of leukemia cells in vitro and in vivo. Np9 not only activated ERK, AKT and Notch1 pathways but also upregulated β-catenin essential for survival of leukemia stem cells. In human leukemia, Np9 protein level in leukemia patients was substantially higher than that in normal donors (56% vs 4.5%). Moreover, Np9 protein level was correlated with the number of leukemia stem/progenitor cells but not detected in normal CD34(+) hematopoietic stem cells. In addition, Np9-positive samples highly expressed leukemia-specific pol-env polyprotein, env and transmembrane proteins as well as viral particles. Thus, the viral oncogene Np9 is a critical molecular switch of multiple signaling pathways regulating the growth of leukemia stem/progenitor cells. These findings open a new perspective to understand the etiology of human common leukemia and provide a novel target for treating leukemia.
CD133 is widely expressed in colorectal cancer (CRC) tissues and cell lines. This protein has been used as a marker of CRC cancer stem cells, although the function and mechanism of CD133 in CRC invasion and metastasis remain unclear. In our study, we examined the role of CD133 in CRC invasion in vitro and investigated the mechanism involved in CD133-related invasion. CD133(high) and CD133(low) HCT116 cells were isolated, and the proliferation and invasive ability of these two subpopulations were tested. CD133(high) HCT116 cells exhibited greater proliferation and invasion compared with CD133(low) HCT116 cells. CD133 knockdown (using CD133 small-interfering [si]RNA) inhibited the invasive activity of CD133si-HCT116 cells. For the first time, we found that the expression of tissue inhibitor of matrix metalloproteinases-2 (TIMP-2) was down-regulated in CD133si-HCT116 cells. In addition, for the TIMP-2si-HCT116 cells (transfected with TIMP-2 siRNA), in vitro invasion was significantly decreased, whereas the expression of CD133 remained unchanged. Finally, the metalloproteinase 2 and MEK/ERK signaling pathways were examined, and no significant change was observed after the knockdown of CD133 or TIMP-2 in HCT116 cells. In conclusion, we demonstrated that CD133 plays an important role in HCT116 cell invasion, and for the first time, we found that CD133 knockdown significantly down-regulated TIMP-2 expression, which suggests that CD133 affects the invasive ability of HCT116 cells by regulating TIMP-2.
Toosendanin (TSN), a triterpenoid isolated from Melia toosendan Sieb. et Zucc., has been found to suppress proliferation and induce apoptosis in a variety of human cancer cells. However, the mechanism how TSN induces apoptosis remains poorly understood. In this study, we examined the effects of TSN on the growth, cell cycle arrest, induction of apoptosis and the involved signaling pathway in human promyelocytic leukemia HL-60 cells. Proliferation of HL-60 cells was inhibited in a dose-dependent manner with the IC(50 (48 h)) of 28 ng/mL. The growth inhibition was due primarily to the S phase arrest and cell apoptosis. Cell apoptosis induced by TSN was confirmed by Annexin V-FITC/propidium iodide staining. The increase of the pro-apoptotic protein Bax, cleaved PARP and caspase-3, and the decrease of anti-apoptotic protein Bcl-2 were observed. Western blot analysis indicated that TSN inhibits the CDC42/MEKK1/JNK pathway. Taken together, our study suggested, for the first time, that the pro-apoptotic effects of TSN on HL-60 cells were mediated through JNK signaling pathway.
This study examined the ability of 1,2,3,4,6-penta-O-galloyl-β-D-glucose (β-PGG) to induce the expression of heme oxygenase-1 (HO-1) in the PC12 cells and its regulation in the PC12 cells. One week before treatment with the drug, nerve growth factor (NGF) was added to the cultures at a final concentration of 50 ng/mL to induce neuronal differentiation. After drug treatment, HO-1 gene transcription was analyzed by reverse transcription polymerase chain reaction (RT-PCR). Expression of HO-1 and NF-E2-related factor2 (Nrf2) and activation of extracellular signal-regulated kinase (ERK) and Akt were detected by Western blotting. The viability of the PC12 cells treated with different medicines was examined by MTT assay. The oxidative stress in the PC12 cells was evaluated qualitatively and quantitatively by DCFH-DA. The results showed that β-PGG up-regulated HO-1 expression and this increased expression provided neuroprotection against MPP(+)-induced oxidative injury. Moreover, β-PGG induced Nrf2 nuclear translocation, which was found to be upstream of β-PGG-induced HO-1 expression, and the activation of ERK and Akt, a pathway that is involved in β-PGG-induced Nrf2 nuclear translocation, HO-1 expression and neuroprotection. In conclusion, β-PGG up-regulates HO-1 expression by stimulating Nrf2 nuclear translocation in an ERK- and Akt-dependent manner, and HO-1 expression by β-PGG may provide the PC12 cells with an acquired antioxidant defense capacity to survive the oxidative stress.
As a potential bioactive material, β-calcium silicate (β-CS) has attracted particular attention in the field of bone regeneration. In this study, porous β-CS/Poly-D,L-Lactide-Glycolide (PDLGA) composite scaffolds were developed with the goals of controlling the degradation rate and improving the mechanical and biological properties. The compressive strength and toughness were significantly enhanced by PDLGA modification of porous β-CS ceramic scaffolds. The effects of the ionic extract from β-CS/PDLGA composite scaffolds on osteogenic differentiation of rat bone marrow-derived mesenchymal stem cells (rBMSCs), proliferation of human umbilical vein endothelial cells (HUVECs) and the related mechanisms were investigated. It was shown that bioactive ions from β-CS/PDLGA scaffolds could enhance cell viability, alkaline phosphatase (ALP) activity, calcium mineral deposition, and mRNA expression levels of osteoblast-related genes of rBMSCs without addition of extra osteogenic reagents. The activation in AMP-activated protein kinase (AMPK), extracellular signal-related kinases (ERK) 1/2 and RUNX-2 were observed in rBMSCs cultured in the extract of β-CS/PDLGA, and these effects could be blocked by AMPK inhibitor Compound C. The extracts of β-CS/PDLGA composites stimulated HUVECs proliferation that was associated with phosphorylation of protein kinase B (Akt) and endothelial nitric oxide synthase (eNOS) as well as an increase in nitric oxide (NO) production and secretion of vascular endothelial growth factor (VEGF). The inductions were abolished by the addition of phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002. The composite scaffolds were implanted in critical sized rabbit femur defects (6 × 10 mm) for 4, 12 and 20 weeks with β-tricalcium phosphate (β-TCP) as controls. Sequential histological evaluations and radiographs revealed that β-CS/PDLGA dramatically stimulated new bone formation and angiogenesis. The biodegradation rate of the β-CS/PDLGA scaffolds was lower than that of β-TCP at each time point examined, and matched the new bone formation rates. These data suggest that β-CS/PDLGA could promote bone regeneration in vivo, which might be ascribed to the enhanced osteogenic differentiation of mesenchymal stem cells (MSCs) and increased angiogenic activity of endothelial cells (ECs).
Increasing evidence suggests that inflammatory microenvironment plays a critical role at different stages of tumor development. However, the molecular mechanisms of the interaction between inflammation and proliferation of cancer cells remain poorly defined. Here we reported the inhibitory effects of oroxylin A on the inflammation-stimulated proliferation of tumor cells and delineated the mechanism of its action. The results indicated that treatment with oroxylin A inhibited NF-κB p65 nuclear translocation and phosphorylation of IκBα and IKKα/β in both human colon tumor HCT116 cells and human monocytes THP-1 cells. In addition, in THP-1 cells, oroxylin A significantly suppressed lipopolysaccharide (LPS)-induced secretion of prototypical proinflammatory cytokine IL-6 but not IL-1β, and it was confirmed at the transcription level. Moreover, oroxylin A inhibited the proliferation of HCT116 cells stimulated by LPS-induced THP-1 cells in co-culture microenvironment. In summary, oroxylin A modulated NF-κB signaling pathway involved in inflammation-induced cancer initiation and progression and therefore could be a potential cancer chemoprevention agent for inflammation-related cancer.
The adhesion of monocytes to activated vascular endothelial cells is a critical event in the initiation of atherosclerosis. Adhesion is mediated by oxidized low-density lipoprotein (ox-LDL) which up-regulates inflammatory markers on endothelial cells. Here we report that (±) 7, 8-dihydroxy-3-methyl-isochromanone-4 (XJP-1), an inhibitor of ox-LDL-induced adhesion of monocytes to endothelial cells blocks cellular functions which are associated with adhesion. We show that XJP-1 down-regulates ox-LDL-induced over-expression of adhesion molecules (ICAM-1 and VCAM-1) in a dose-dependent manner in human umbilical vein endothelial cells (HUVECs), attenuates ox-LDL-induced up-regulation of low-density lipoprotein receptor (LOX)-1, decreases generation of reactive oxygen species (ROS), blocks translocation of nuclear factor-kappa B (NF-κB) activity, and prevents activation of c-Jun N-terminal kinase (JNK)/p38 pathways in endothelial cells. These findings suggest that XJP-1 may attenuate ox-LDL-induced endothelial adhesion of monocytes by blocking expression of adhesion molecules through suppressing ROS/NF-κB, JNK and p38 pathways.
In this study, the effect of heparin-derived oligosaccharide on bovine vascular smooth muscle cell (VSMC) proliferation and signal transduction mechanism was investigated. Extracellular-signal-regulated kinase (ERK) 1/2 has been implicated in the regulation of various cellular functions including proliferation, and we sought to define a functional role for ERK 1/2 in an established proliferation model in order to find a possible mechanism for inhibition of VSMC proliferation by heparin-derived oligosaccharide. The VSMC proliferation model was developed by platelet-derived growth factor (PDGF), and the level of ERK 1/2 protein and messenger RNA was determined by reverse transcriptase-polymerase chain reaction, Western blotting, and immunocytochemical methods. Flow cytometry analysis indicated that heparin-derived oligosaccharide blocked PDGF-induced cell cycle progression by arresting cells in the G0/G1 phase. The results imply that heparin-derived oligosaccharide inhibits VSMC proliferation by moderating the gene and the phosphorylation levels of ERK 1/2, eventually blocking G1/S transition, may be one of the mechanisms for inhibition of VSMC proliferation by heparin-derived oligosaccharide.
It has been well characterized that flavonoids possess pronounced anticancer potentials including anti-angiogenesis, anti-metastasis, and pro-apoptosis. Herein, we report, for the first time, that VI-14, a novel flavonoid derivative, possesses anti-cancer properties. The purpose of this study is to investigate the anti-migration and anti-invasion activities of VI-14 in breast cancer cells. Our data indicate that VI-14 inhibits adhesion, migration and invasion of MDA-MB-231 and MDA-MB-435 human breast cancer cells. MDA-MB-231 cells treated with VI-14 display reduced activities and expressions of ECM degradation-associated proteins including matrix metalloproteinase 2 (MMP-2) and 9 (MMP-9) at both the protein and mRNA levels. Meanwhile, VI-14 treatment induces an up-regulated expression of tissue inhibitor of metalloproteinase 1 (TIMP-1) and 2 (TIMP-2) in MDA-MB-231 cells. Western blotting results show that phosphorylation levels of critical components of the MAPK signaling pathway, including ERK, JNK and P38, are dramatically decreased in VI-14-treated MDA-MB-231 cells. Furthermore, treatment of VI-14 significantly decreases the nuclear levels and the binding ability of nuclear factor-kappa B (NF-κB) and activator protein-1 (AP-1). Taken together, our data suggest that VI-14 treatment suppresses migration and motility of breast cancer cells, and VI-14 may be a potential compound for cancer therapy.
Cardiac hypertrophy is a response of the myocardium to increased workload and is characterised by an increase of myocardial mass and an accumulation of extracellular matrix (ECM). As an ECM protein, an integrin ligand, and an angiogenesis inhibitor, all of which are key players in cardiac hypertrophy, mindin is an attractive target for therapeutic intervention to treat or prevent cardiac hypertrophy and heart failure. In this study, we investigated the role of mindin in cardiac hypertrophy using littermate Mindin knockout (Mindin ( -/- )) and wild-type (WT) mice. Cardiac hypertrophy was induced by aortic banding (AB) or angiotensin II (Ang II) infusion in Mindin ( -/- ) and WT mice. The extent of cardiac hypertrophy was quantitated by echocardiography and by pathological and molecular analyses of heart samples. Mindin ( -/- ) mice were more susceptible to cardiac hypertrophy and fibrosis in response to AB or Ang II stimulation than wild type. Cardiac function was also markedly exacerbated during both systole and diastole in Mindin ( -/- ) mice in response to hypertrophic stimuli. Western blot assays further showed that the activation of AKT/glycogen synthase kinase 3β (GSK3β) signalling in response to hypertrophic stimuli was significantly increased in Mindin ( -/- ) mice. Moreover, blocking AKT/GSK3β signalling with a pharmacological AKT inhibitor reversed cardiac abnormalities in Mindin ( -/- ) mice. Our data show that mindin, as an intrinsic cardioprotective factor, prevents maladaptive remodelling and the transition to heart failure by blocking AKT/GSK3β signalling.
Keloid is a specific skin scar that expands beyond the boundaries of the original injury as it heals. The invasive nature of keloid and notable migratory activity of fibroblasts are a hallmark, which distinguishes keloids from other common scars. Madecassoside, a triterpenoid saponin occurring in Centella asiatica herbs, possesses unique pharmacological properties to enhance wound-healing and diminish keloid formation. However, the effects of madecassoside on the formation of keloid scars have been poorly understood. Here, we focused on the potential of madecassoside on the migration of keloid-derived fibroblasts (KFs) and its mechanism. Primary KF, originating from human earlobe keloids, were purified and cultured, and then treated with madecassoside (10, 30, and 100μM). In both transwell migration assays and scratch-wound-closure assays, KF migration was considerably suppressed by madecassoside pretreatment. Furthermore, KFs treated with madecassoside showed decreased F-actin filaments, as revealed by fluorescein isothiocyanate (FITC)-phalloidin staining and confocal microscopy. By Western blot analysis, madecassoside was shown to remarkably attenuate the phosphorylation of cofilin, p38 MAPK and phosphatidylinositol-3-kinase (PI3K)/AKT signaling, but only exhibited a minor effect on MMP-13 and little effect on ERK1/2 phosphorylation. It was concluded that madecassoside could be of great use in the treatment and/or prevention of hypertrophic scars and keloids.
A tightly regulated catabolic process named autophagy involves the degradation of intracellular components via lysosomes. Here we investigate the antitumor effect of E Platinum, a newly synthesized derivative of oxaliplatin, in vivo and in vitro. E Platinum exhibits growth inhibition of various tumor cells in a dose-dependent manner, but the mechanism underlying it is unclear. Based on theory introducing autophagy, we preliminarily investigate whether autophagy could contribute to the antitumor activity of E Platinum. Our results showed that autophagy induced by 12.5 μM E Platinum in gastric carcinoma BGC-823 cells was significantly characterized by the FITC-fluorescent microtubule associated protein 1 light chain 3 (MAP-LC3), lysosomal-rich/acidic compartments visualized with Lysotracker red (LTR-red) and an accumulation of numerous large autophagic vesicles within the cytoplasm, but not in the control cells. Meanwhile treatment of cells with 12.5 μM E Platinum resulted in conversion of water soluble LC3 (LC3-I) to lipidated and autophagosome-associated form (LC3-II) as well as increasing expression of autophagy protein Beclin 1. Activation of predominant lysosomal aspartic protease, LAMP-1 and cathepsin D, was demonstrated. Moreover, RNA interference targeting Beclin 1, inhibition of autophagy by 3-methyladenine (3-MA) and chloroquine significantly suppressed the above process as well as the BGC-823 cells growth inhibition triggered by 12.5 μM E Platinum. Studies of mechanism revealed that E Platinum suppressed activation of mTOR and p70S6K by decreasing phosphorylation of Akt, ERK1/2, JNK and p38 involved in mitogen-activated protein kinase signaling. We supported new evidences for E Platinum as a promising antitumor agent, involving with autophagy induction.
An experiment was performed to observe the changes in Raf-1 kinase/mitogen-activated protein kinase ERK (MEK)/extracellular signal-regulated kinase (ERK) signaling pathways in cultured hippocampal neurons and its correlation with neurons apoptosis induced by intracellular zinc depletion. Cultured hippocampal neurons were exposed to a cell membrane-permeant zinc chelator TPEN (2 µM), and to TPEN plus zinc sulfate (5 µM) for 24 h. Cultures were then processed to detect neuronal viability by the methyl thiazolyl tetrazolium assay, while apoptosis rate was simultaneously observed by the flow cytometric analysis. Caspase-3, Raf-1, pMEK, pERK1/2, and pCREB protein levels were examined by Western blot assays. The viability in TPEN-incubated neurons was notably decreased, apoptosis rate and expression of caspase-3 significantly increased compared to untreated controls. The significant down-regulation of Raf/MEK/ERK signaling pathway and expression of pCREB were decreased in TPEN-treated neurons. Co-addition of zinc almost completely reversed TPEN-induced alterations described. The results demonstrated zinc-modulated apoptosis and the expression of Raf/MEK/ERK at the protein level in hippocampal neurons. It is possible that zinc depletion-induced apoptosis in cultured hippocampal neurons may be relevant to the changes of Raf/MEK/ERK signaling pathway.
Flavonoid is an important group of natural products which exerted anticancer effects against various cancers. III-10 is a newly synthesized flavonoid with a pyrrolidinyl and a benzyl group substitution. It shares a same carbon skeleton with flavonoid which indicates that it may also have potential anticancer activity. To investigate whether III-10 could express anticancer effect on human U937 leukemia cells through differentiation induction, a series of experiments were processed. MTT and trypan-blue excluding assays showed that III-10 possessed growth and viability inhibition effects on U937 cells. Giemsa staining was implemented to observe the morphologic changes of U937 cells after III-10 treatment. Nitroblue tetrazolium (NBT) reduction assay and cell surface antigen flow cytometry were carried out to figure out the way III-10 induced U937 cells to differentiate. These experiments revealed that III-10 could induce U937 cells to differentiate into monocyte-like cells. Western blots and immunofluorescence were performed to inspect the possible underlying mechanism. The results revealed that differentiation-related proteins phospholipids scramblase 1 (PLSCR1) and promyelocytic leukemia protein (PML) were up-regulated after III-10 treatment. And III-10 stimulated PLSCR1 and PML probably through activation of protein kinase Cδ (PKCδ). All these results suggested that III-10 was a prospective anticancer compound and was requisite to be proceeded further investigation.
Matrix metalloproteinases (MMPs) play important roles in the invasion and migration of cancer cells. In this study, we used in vitro and in vivo assays to examine the inhibitory effects of oroxylin A, one of the main bioactive flavonoid extracted from Scutellaria radix, on the human breast carcinoma cell MDA-MB-231 invasion and migration. We found that oroxylin A can suppress cell adhesion, invasion and migration in a concentration-dependent manner. Moreover, oroxylin A led to the reduction of the activity and expression levels of MMP-2 and MMP-9 in gelatin zymography, real-time PCR and western blotting analysis. Further elucidation of the mechanism revealed that oroxylin A increased the expression of tissue inhibitor of metalloproteinase-2 (TIMP-2), the endogenous inhibitor of MMP-2, and repressed the phorbol-12-myristate-13-acetate (PMA)-induced translocation of protein kinase Cδ (PKCδ), phosphorylation of extracellular signal-regulated kinase (ERK1/2) and binding activity of the transcription factor activator protein-1 (AP-1) which are upstream signaling molecules in MMP-9 expression. Our results also indicated that oroxylin A inhibited the lung metastasis of murine melanoma cell B16-F10 in vivo. Therefore, we proposed that oroxylin A might be developed as a therapeutic potential candidate for the treatment of cancer metastasis.
Insulin-like growth factor 1 (IGF-1) is a potent mitogenic protein which can enhance the osteogenic differentiation of periodontal ligament (PDL) fibroblasts. However, it remains unclear whether IGF-1 can stimulate the osteogenic differentiation and osteogenesis of human periodontal ligament stem cells (PDLSCs). In this study, STRO-1(+) PDLSCs were isolated from human PDL tissues, treated with IGF-1, and their osteogenic capacity was investigated in vitro and in vivo. Dimethyl-thiazol-diphenyl tetrazolium bromide assay and flow cytometry results demonstrated that 10-200 ng/mL IGF-1 can stimulate the proliferation ability of PDLSCs and 100 ng/mL is the optimal concentration. Exogenous IGF-1 can modify the ultrastructure, enhance the alkaline phosphatase activity, the mineralization ability of PDLSCs, and increase the expression of osteogenic markers (runt-related transcription factor 2, osterix, and osteocalcin) at mRNA and protein levels. In vivo transplantation illustrated that IGF-1 treated implants generated more mineralized tissues, and presented stronger expression of RUNX2, OSX, and OCN than control group. Moreover, the expression of phosphor-ERK and phosphor-JNK in these stem cells was upregulated by IGF-1, indicating that MAPK signaling pathway was activated during the osteogenic differentiation of PDLSCs mediated by IGF-1. Together, the results showed that IGF-1 can promote the osteogenic differentiation and osteogenesis of STRO-1(+) PDLSCs via ERK and JNK MAPK pathway, suggesting that IGF-1 is a potent agent for stem cell-based periodontal tissue regeneration.
It has been known that Rho-associated protein kinase (ROCK) signaling regulates the migration of vascular smooth muscle cells (VSMCs). However, the isoform-specific roles of ROCK and its underlying mechanism in VSMC migration are not well understood. The current study thus aimed to investigate the roles of ROCK1/2 and their relationship to the MAPK signaling pathway in platelet-derived growth factor (PDGF)-induced rat aorta VSMC migration by manipulating ROCK gene expression. The results revealed that ROCK1 small interfering ribonucleic acid (siRNA) rather than ROCK2 siRNA decreased PDGF-BB-generated VSMC migration, and upregulation of ROCK1 expression via transfection of constructed pEGFP-C1/ROCK1 plasmid further increased the migration of PDGF-BB-treated VSMCs. In PDGF-treated VSMCs, ROCK1 siRNA did not affect the phosphorylation levels of ERK and p38 in the cytoplasm, but decreased the level of ERK phosphorylation in the nucleus. These findings demonstrate that activated ROCK1 can promote VSMC migration through facilitating phosphorylation and nuclear translocation of ERK protein.
Metastasis of tumor cells is associated with epithelial-to-mesenchymal transition (EMT), which is a process whereby epithelial cells lose their polarity and acquire new features of mesenchyme. EMT has been reported to be induced by transforming growth factor-β1 (TGF-β1), but its mechanism remains elusive. In this study, we performed a study to investigate whether PI3K/Akt and MAPK/Erk1/2 signaling pathways involved in EMT in the human lung cancer A549 cells. The results showed that after treated with TGF-β1 for 48 h, A549 cells displayed more fibroblast-like shape, lost epithelial marker E-cadherin and increased mesenchymal markers Vimentin and Fibronectin. Moreover, TGF-β1-induced EMT after 48 h was accompanied by increased of cell migration and change of Akt and Erk1/2 phosphorylation. In addition, EMT was reversed by PI3K inhibitor LY294002 and MEK1/2 inhibitor U0126, which suggested that A549 cells under stimulation of TGF-β1 undergo a switch into mesenchymal cells and PI3K/Akt and MAPK/Erk1/2 signaling pathways serve to regulate TGF-β1-induced EMT of A549 cells.
We examined the effects of anti-six-transmembrane epithelial antigen of the prostate-4 (STEAP4) antibodies on glucose transport in mature adipocytes and determined the mechanism of insulin resistance in obesity. Western blotting was performed to determine STEAP4 expression, to assess translocation of insulin-sensitive glucose transporter 4 (GLUT4), and to measure phosphorylation and total protein content of insulin-signaling proteins. Confocal laser microscopy and flow cytometry were used to detect intracellular reactive oxygen species (ROS) and fluctuations in mitochondrial membrane potential (ΔΨ). ATP production was measured by using a luciferase-based luminescence assay kit. After the application of anti-STEAP4 antibodies at 0.002 mg/mL, adipocytes exhibited reduced insulin-stimulated glucose transport by attenuating the phosphorylation of IRS-1, PI3K (p85), and Akt. The antibodies also potentially increase the level of ROS and decrease cellular ATP production and ΔΨ. In conclusion, (i) STEAP4 regulates the function of IRS-1, PI3K, and Akt and decreases insulin-induced GLUT4 translocation and glucose uptake; (ii) ROS-related mitochondrial dysfunction may be related to a reduced IRS-1 correlation with the PI3K signaling pathway, leading to insulin resistance. These observations highlight the potential role of STEAP4 in glucose homeostasis and possibly in the pathophysiology of type 2 diabetes related to obesity and may provide new insights into the mechanisms of insulin resistance in obesity.
The molecular mechanisms underlying the behavioral effects of glucocorticoids are poorly understood. We report here that hippocampal neuronal nitric oxide synthase (nNOS) is a crucial mediator. Chronic mild stress and glucocorticoids exposures caused hippocampal nNOS overexpression via activating mineralocorticoid receptor. In turn, hippocampal nNOS-derived nitric oxide (NO) significantly downregulated local glucocorticoid receptor expression through both soluble guanylate cyclase (sGC)/cGMP and peroxynitrite (ONOO(-))/extracellular signal-regulated kinase signal pathways, and therefore elevated hypothalamic corticotrophin-releasing factor, a peptide that governs the hypothalamic-pituitary-adrenal axis. More importantly, nNOS deletion or intrahippocampal nNOS inhibition and NO-cGMP signaling blockade (using NO scavenger or sGC inhibitor) prevented the corticosterone-induced behavioral modifications, suggesting that hippocampal nNOS is necessary for the role of glucocorticoids in mediating depressive behaviors. In addition, directly delivering ONOO(-) donor into hippocampus caused depressive-like behaviors. Our findings reveal a role of hippocampal nNOS in regulating the behavioral effects of glucocorticoids.
We investigated the mechanisms underlying the protective effects of loganin against hydrogen peroxide (H(2)O(2))-induced neuronal toxicity in SH-SY5Y cells. The neuroprotective effect of loganin was investigated by treating SH-SY5Y cells with H(2)O(2) and then measuring the reduction in H(2)O(2)-induced apoptosis using 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and lactate dehydrogenase (LDH) release assays. Following H(2)O(2) exposure, Hoechst 33258 staining indicated nuclear condensation in a large proportion of SH-SY5Y cells, along with an increase in reactive oxygen species (ROS) production and an intracellular decrease in mitochondria membrane potential (MMP). Loganin was effective in attenuating all the above-stated phenotypes induced by H(2)O(2). Pretreatment with loganin significantly increased cell viability, reduced H(2)O(2)-induced LDH release and ROS production, and effectively increased intracellular MMP. Pretreatment with loganin also significantly decreased the nuclear condensation induced by H(2)O(2). Western blot data revealed that loganin inhibited the H(2)O(2)-induced up-regulation of cleaved poly (ADP-ribose) polymerase (PARP) and cleaved caspase-3, increased the H(2)O(2)-induced decrease in the Bcl-2/Bax ratio, and attenuated the H(2)O(2)-induced release of cytochrome c from mitochondria to the cytosol. Furthermore, pretreatment with loganin significantly attenuated the H(2)O(2)-induced phosphorylation of c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase (MAPK), and extracellular signal-regulated kinase 1/2 (ERK 1/2). These results suggest that the protective effects of loganin against H(2)O(2)-induced apoptosis may be due to a decrease in the Bcl-2/Bax ratio expression due to the inhibition of the phosphorylation of JNK, p38, and ERK 1/2 MAPKs. Loganin's neuroprotective properties indicate that this compound may be a potential therapeutic agent for the treatment of neurodegenerative diseases.
We investigated the neuroprotective effects of Lonicera japonica THUNB. (Caprifoliaceae) (LJ) extract against hydrogen peroxide (H(2)O(2)), a toxin created by oxidative stress and implicated in neurodegenerative diseases, in human SH-SY5Y neuroblastoma cells. We examined the effects of LJ against H(2)O(2)-induced cytotoxicity, apoptosis, the production of reactive oxygen species (ROS), the proteolysis of cleaved poly-ADP-ribose polymerase (PARP), and the expression of Bcl-2, Bcl-xL, and cleaved caspase-3. Moreover, we attempted to determine whether LJ suppressed the phosphorylation of Akt, c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase (MAPK), and extracellular signal-regulated kinase 1/2 (ERK 1/2). We found that LJ improved cell viability, inhibited cytotoxicity and apoptosis, and attenuated elevations in ROS and nuclear condensation. In addition, LJ showed radical scavenging ability in 2-diphenyl-1-picrylhydrazyl (DPPH) and 2,2-azinobis-(3-ethyl-benzthiazoline-6-sulfonic acid) (ABTS) assays. Western blot data revealed that LJ inhibited H(2)O(2)-induced up- and down-regulation of cleaved PARP, cleaved caspase-3, Bcl-2, and Bcl-xL. Furthermore, LJ significantly attenuated the H(2)O(2)-induced phosphorylation of Akt, JNK, p38 MAPK, and ERK1/2. These results demonstrate that LJ possesses potent neuroprotective activity. Its potential to treat neurodegenerative diseases warrants further research.
Oxidative stress is a major cause in neurodegenerative diseases including Alzheimer's disease (AD), Parkinson's disease (PD), and cerebral ischemia. Ginsenoside Rg1, a natural product extracted from Panax ginseng C.A. Meyer, has been reported to exert notable neuroprotective activities, which partly ascribed to its antioxidative activity. However, its molecular mechanism against oxidative stress induced by exogenous hydrogen peroxide (H(2)O(2)) remained unclear. In this study, we investigated its effect on H(2)O(2)-induced cell death and explored possible signaling pathway in PC12 cells. We proved that pretreatment with Rg1 at concentrations of 0.1-10 μM remarkably reduced the cytotoxicity induced by 400 μM of H(2)O(2) in PC12 cells by MTT and Hoechst and PI double staining assay. Of note, we demonstrated the activation of NF-κB signaling pathway induced by H(2)O(2) thoroughly in PC12 cells, and Rg1 suppressed phosphorylation and nuclear translocation of NF-κB/p65, phosphorylation and degradation of inhibitor protein of κB (IκB) as well as the phosphorylation of IκB-kinase complex (IKK) by western blotting or indirect immunofluorescence assay. Besides, Rg1 also inhibited the activation of Akt and the extracellular signal-regulated kinase 1/2 (ERK1/2). Furthermore, the protection of Rg1 on H(2)O(2)-injured PC12 cells was attenuated by pretreatment with two NF-κB pathway inhibitors (JSH-23 or BOT-64). In conclusion, our results suggest that Rg1 could rescue the cell injury by H(2)O(2) via down-regulation NF-κB signaling pathway as well as Akt and ERK1/2 activation, which put new evidence on the neuroprotective mechanism of Rg1 against the oxidative stress and the regulatory role of H(2)O(2) in NF-κB pathway in PC12 cells.
Drug carriers are generally introduced into the body intravenously and directly exposed to endothelial cells. Silica nanoparticles could be promising delivery vehicles for drug targeting or gene therapy. However, few studies have been undertaken to determine the biological behavior of silica nanoparticles on endothelial cells. Here we measured reactive oxygen species (ROS) generation, apoptosis and necrosis, proinflammatory and prothrombic properties and the levels of the apoptotic signaling proteins and the transcription factors in human umbilical vein endothelial cells (HUVECs) after exposure to silica nanoparticles of different concentrations (25, 50, 100, and 200 microg/mL) for 24h. The results showed that silica nanoparticles, ranging from 50 microg/mL to 200 microg/mL, markedly induced ROS production, mitochondrial depolarization and apoptosis in HUVECs. At the highest concentration, the necrotic rate, LDH leakage, the expression of CD54 and CD62E, and the release of TF, IL-6, IL-8 and MCP-1 were significantly increased. Silica nanoparticles also activated c-Jun N-terminal kinase (JNK), c-Jun, p53, caspase-3 and NF-kappaB, increased Bax expression and suppressed Bcl-2 protein. Moreover, inhibition of ROS attenuated silica nanoparticles-induced apoptosis and inflammation and the activation of JNK, c-Jun, p53 and NF-kappaB. In summary, our findings demonstrated that silica nanoparticles could induce dysfunction of endothelial cells through oxidative stress via JNK, p53 and NF-kappaB pathways, suggesting that exposure to silica nanoparticles may be a significant risk for the development of cardiovascular diseases such as atherosclerosis and thrombus.
Gambogic acid (GA) has been known to have antitumor activity in vitro and in vivo. In the present study, we investigated the anti-invasive effects of GA in MDA-MB-231 human breast carcinoma cells. The results indicated that GA significantly inhibited the adhesion, migration, and invasion of the cells in vitro tested by the heterotypic adhesion assay, wound migration assay, and chamber invasion assay. Results of Western blotting and immunocytochemistry analysis showed that GA could suppress the expressions of matrix metalloproteinase 2 (MMP-2) and 9 (MMP-9) in MDA-MB-231 cells. Furthermore, gelatin zymography revealed that GA decreased the activities of MMP-2 and MMP-9. Additionally, GA exerted an inhibitory effect on the phosphorylation of ERK1/2 and JNK, while it had no effect on p38. Taken together, our results demonstrated the anti-invasive property of GA for the first time and indicated it could serve as a promising drug for the treatment of cancer metastasis.
SCOPE:
Flavonoids have well-known antioxidant, anti-inflammatory, and anti-cancer activities. Isoflavone genistein is considered a potent antioxidant agent against oxidative stress. Although several mechanisms have been proposed, a clear antioxidant mechanism of genistein is still remained to be answered.
METHODS AND RESULTS:
In this study, we focused on the concerted effects on expression of Nrf2 and phase II enzyme pathway components. Transient transfection assays, RT-PCR and immunoblot analysis were performed to study its molecular mechanisms of action. In Caco-2 cells, treatment with genistein markedly attenuated H(2)O(2) -induced peroxide formation; this amelioration was reversed by buthionine sulfoximine(GCLC inhibitor) and zinc protoporphyrin(HO-1 inhibitor). Genistein increased HO-1 and GCLC mRNA and protein expression. Genistein treatment activated the ERK1/2 and PKC signaling pathway; therefore increased Nrf2 mRNA and protein expression. The roles of the ERK1/2 and PKC signaling pathway were determined using PD98059 (ERK1/2 inhibitor) and GF109203X (PKC inhibitor) and RNA interference directed against Nrf2. Both inhibitors and siNrf2 abolished genistein-induced HO-1 and GCLC protein expression. These results suggest the involvement of ERK1/2, PKC, and Nrf2 in inducing HO-1 and GCLC by genistein.
CONCLUSION:
Our studies show that genistein up-regulated HO-1 and GCLC expression through the EKR1/2 and PKC /Nrf2 pathways during oxidative stress.
© 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
BACKGROUND:
There is an obvious relationship among angiogenesis and inflammation. From previous study, we learn that oroxylin A possesses anti-angiogenic activity in vitro and in ovo. It also has an inhibitory effect on inflammation. But whether oroxylin A suppresses the inflammation-induced angiogenesis is still unknown. Our present study focuses on the role of oroxylin A in targeting LPS-induced angiogenesis, inflammatory and related pathways.
METHODS:
The effects of oroxylin A on angiogenesis were investigated by transwell assay, tube formation assay, rat aortic ring assay and chorioallantoic membrane (CAM) model. Western blotting analysis was used to detect the expression of certain proteins.
RESULTS:
We found that oroxylin A inhibited LPS-induced migration and tube formation of human umbilical vein endothelial cells (HUVECs), as well as microvessel sprouting from rat aotric ring in vitro and the angiogenesis of chicken chorioallantoic membrane (CAM) model in ovo. The results also indicated that oroxylin A could inhibit the expression of LPS acceptor toll-like receptor 4 (TLR4) and the activities of its downstream mitogen-activated protein kinases (MAPKs), including reducing expressions of the phosphorylation of JNK, p38, and ERK. Moreover, oroxylin A prevented NF-κB dimers from translocating to the nucleus.
CONCLUSIONS:
Taken together, oroxylin A can suppress the angiogenesis induced by LPS and it may affect the LPS/TLR4 signaling pathway.
OBJECTIVE:
MicroRNA-133a (miR-133a) and microRNA-133b (miR-133b) are located on chromosome 18 in the same bicistronic unit. Recently, they have been commonly identified as being down-regulated in various human malignancies, such as bladder cancer, pancreatic ductal adenocarcinoma, oesophageal squamous cell carcinoma of the tongue, and hepatocellular and lung carcinomas. The present study examined the effects of miR-133a and miR-133b in bladder cancer T24 and EJ cells.
MATERIAL AND METHODS:
After transfection of miR-133a and miR-133b, the expression of miR-133a/b was assessed, and a cell viability assay, cell migration assay, cell invasion assay, luciferase assay and Western blot were conducted in bladder cancer T24 and EJ cells.
RESULTS:
Both miR-133a and miR-133b were found to inhibit cell proliferation, migration and invasion in T24 and EJ cells. The first evidence was provided that miR-133a and miR-133b may directly target the epidermal growth factor receptor in bladder cancer.
CONCLUSIONS:
This study provided the first glimpse of the functional role of miR-133 in bladder cancer T24 and EJ cells. The results may increase our knowledge on the molecular basis of progression and provide potential therapy for bladder cancer.
PURPOSE:
In this study, the effect of heparin-derived oligosaccharide (HDO) on vascular endothelial growth factor (VEGF) induced vascular smooth muscle cell (VSMC) proliferation and the signal transduction mechanisms involved were investigated.
METHODS:
MTT assays were used to measure VSMC proliferation, flow cytometry to analyze cell cycle distribution, RT-PCR for detection of gene transcript levels, and cell-based ELISA, Western blotting and immunocytochemical methods to detect the expression of PKC-α, ERK 1/2, p-ERK 1/2, Akt, p-Akt, p-PDK1 and p-GSK-3β.
RESULTS:
HDO at concentrations of 0.01, 0.1 and 1 μmol·L(-1) dose-dependently inhibited VEGF-induced VSMC proliferation with inhibition indices of 6.8 %, 13.1 % and 28.9 %, respectively. Similar concentrations of HDO dose-dependently decreased the percentage of VEGF-induced cells in S phase to 3.6 %, 3.4 %, and 5.4 %, while increasing that of cells arrested in the G0/G1 phase to 80 %, 82 % and 83.6 %. HDO at 0.01, 0.1 or 1 μmol·L(-1) inhibited VEGF-induced PKC-α mRNA expression, with inhibition indices of 9.2 %, 16.1 % and 54.0 %. HDO at 0.1 or 1 μmol·L(-1) inhibited VEGF-induced proto-oncogene mRNA expression, with inhibition indices of 5.2 % and 6.6 % for c-jun, 8.8 % and 11.6 % for c-myc, and 6.5 % and 11.9 % for c-fos, respectively. Additionally, treatment with 0.01, 0.1 or 1 μmol·L(-1) HDO, inhibited VEGF-induced expression of some proliferation related proteins with inhibition indices of 33.2 %, 56.3 % and 77.0 % for PKC-α, 33.7 %, 38.7 % and 53.2 % for p-Akt, 3.5 %, 24.2 % and 49.3 % for p-ERK 1/2, 39.2 %, 71.8 % and 80.7 % for p-PDK 1 and 41.4 %, 89.4 % and 92.4 % for p-GSK-3β, respectively. The results showed that HDO inhibited PKC-α, c-jun, c-fos and c-myc mRNA transcription, and also down-regulated phosphorylation levels of ERK 1/2 and Akt.
CONCLUSION:
Our study demonstrates that HDO inhibits transcription of proliferation-related proto-oncogenes and arrests G1/S transition through inhibition of the PKC, MAPK and Akt/PI3K pathways in association with inhibition of VSMC proliferation. This altered molecular signature may explain one mechanism of HDO-mediated inhibition of VSMC proliferation.
AIM:
Our previous investigation demonstrated that plasminogen activator inhibitor-1 (PAI-1) siRNA ameliorated bleomycin (BLM)-induced rat lung fibrosis. The present study was undertaken to explore the effect and the mechanism of PAI-1 siRNA and plasmid pcDNA on the proliferation and apoptosis of cultured fibroblasts from BLM-induced fibrotic lung tissues.
MATERIALS AND METHODS:
The fibroblasts from BLM-induced fibrotic lung tissue were isolated and transfected using PAI-1 siRNA and plasmid pcDNA-PAI-1. The techniques of real time RT-PCR and/or western blot were used to determine the expression of PAI-1, α-smooth muscle actin (α-SMA) (real time RT-PCR only), collagen type-1 and type-3 (real time RT-PCR only), and the levels of caspase-3, ERK and AKT signal molecules. The proliferation of fibroblasts was measured by cell cycle with flow cytometry. The intracellular concentration of Ca(2+) was examined by confocal laser microscopy.
RESULTS:
PAI-1 siRNA downregulated the PAI-1 mRNA expression by 70%±7% at 24h and protein expression by 73.5%±10% and 42%±3% at 48h and 72h compared to Non-specific siRNA group. Flow cytometry showed that the fibroblasts at the G(2)M+S phase were significantly reduced by 20.56±1.03% after transfecting PAI-1 siRNA and were significantly increased by 43.8±1.21% after transfecting plasmid pcDNA-PAI-1. The mRNA expressions of α-SMA, collagen type-1and type-3 were downregulated after transfecting the PAI-1 siRNA, while upregulated after the transfection of pcDNA-PAI-1. PAI-1 siRNA increased the level of caspase-3, inhibited the expressions of p-ERK and p-AKT protein molecules, while the pcDNA-PAI-1 transfection showed a reversal effect on these expressions. Intracellular Ca(2+) concentration was decreased after transfecting PAI-1 siRNA, whereas increased after transfecting pcDNA-PAI-1.
CONCLUSION:
PAI-1 promotes the proliferation, transforming into myofibroblasts, collagen synthesis, and inhibits apoptosis of pulmonary fibroblasts by activating Ca(2+), ERK and AKT signaling pathway. Decreasing PAI-1 expression is an available strategy in inhibiting the progression of pulmonary fibrosis.
Copyright © 2012 Elsevier Ltd. All rights reserved.
BACKGROUND:
Castration-resistant prostate cancer (CRPC) is a leading cause of cancer-related deaths in elder men. This disease has limited therapeutic options and poor prognosis as the underlying molecular mechanisms are not clearly understood. Given the emerging roles of microRNA (miRNA) as a key regulator, we postulated that miRNA may play a significant role in CRPC formation.
METHODS:
miR-146a levels in 15 androgen-dependent prostate cancer (ADPC) tissues and 5 CRPC tissues were measured by qRT-PCR. Effects of miR-146a in cell proliferation and migration in vitro and in vivo were evaluated by MTT assay, colony formation assay, transwell migratory assay, and tumor formation assay, respectively.
RESULTS:
we found that miR-146a expression was significantly decreased in CRPC tissues compared to ADPC tissues. Functional analyses showed that ectopic overexpression of miR-146a in androgen-independent cell lines not only inhibited cell growth, colony formation, and migration in vitro, but also reduced tumorigenicity and angiogenesis in vivo. Mechanistic studies revealed that miR-146a repressed the expression of EGFR through binding to its 3'-untranslated region. Also, miR-146a inhibited the expression of MMP2, one of the most important genes in tumor progression. Moreover, downregulation of p-ERK expression significantly abrogated miR-146a-induced prostate cancer cell proliferation.
CONCLUSIONS:
Our findings suggest that ubiquitous loss of miR-146a is a critical mechanism for overexpression of EGFR in CRPC, which is crucial to better understanding the pathogenesis of CRPC.
Copyright © 2011 Wiley Periodicals, Inc.
AIM:
We investigated whether 8-dihydroxy-3-methyl-isochromanone (XJP-1), a novel angiotensin-converting enzyme inhibitor (ACEI), exhibited inhibitory activity to lipopolysaccharide (LPS)-accelerated vascular inflammation.
METHODS:
Human umbilical vein endothelial cells (HUVECs) were isolated from human umbilical cords and cultured. The direct effect of XJP-1 on the activation of endothelial cells was measured using MTT assay. Nitric oxide (NO) in the culture medium was measured using Griess method. The expression of cell adhesion molecules (ICAM-1 and VCAM-1) was determined by flow cytometry and RT-PCR. The protein expression levels of tumor necrosis factor-α (TNF-α), monocyte chemotactic protein (MCP)-1, and endothelin-1 (ET-1) secretion were measured using ELISA. Quantitative analysis of eNOS, iNOS, inhibitory factor NF-κB (IκB) and MAPKs were determined using Western blot analysis. The translocation of NF-κB from the cytoplasm to the nucleus was determined using immunofluorescence.
RESULTS:
XJP-1 significantly inhibited LPS-mediated endothelial cell dysfunction, as measured by NO production, iNOS expression, adhesion molecule (ICAM-1, VCAM-1) expression, and chemokine (TNF-α, MCP-1) production in vitro. It up-regulated eNOS expression in the same experimental setting. XJP-1 alone was found non-cytotoxic at the concentration up to 1000μM. The mechanistic investigations of XJP-1 suppression LPS-induced inflammation in HUVECs revealed that XJP-1 blocked NF-κB nuclear entry in an IκB-dependent manner, as well as inhibited MAPK activation induced by LPS. XJP-1 reduced endothelin-1 secretion and increased nitric oxide metabolite production by HUVECs. However, the effect of XJP-1 on nitric oxide and endothelin-1 metabolite production is mediated by the activation of bradykinin B(2) receptor being counteracted, at least in part, by a specific antagonist.
CONCLUSION:
XJP-1 inhibited LPS-induced cytotoxicity and inflammatory response. The mechanism underlying this protective effect might be related to the inhibition of MAPK and NF-κB signaling pathway activation, suggesting the potential inhibition of the atherosclerotic process by suppressing the expression of chemoattractant molecules and monocyte adhesion. XJP-1 also has an effect in improving endothelin-1 through activating bradykinin B(2) receptor. These findings indicated that XJP-1 is potentially a novel therapeutic candidate for the treatment of atherosclerosis.
Copyright © 2011 Elsevier Ireland Ltd. All rights reserved.
BACKGROUND:
Stimulation of Toll-like receptors (TLRs) by microbial products has been utilised to potentiate immune responses against haematologic malignancies. The maltose-binding protein (MBP) of Escherichia coli could induce the activation of immune cells via TLR4. The aim of the present study was to investigate whether TLRs mediated the biological effects of MBP on U937 and Jurkat cells in vitro. METHODS We observed the effect of MBP on U937 and Jurkat cells by using the WST, cell cycle analysis and morphological observation. Further, cells were stimulated with MBP for indicated times and doses, and detected by RT-PCR, western blotting, immunohistochemistry and immunofluorescence staining to investigate the mechanisms involved in cell viability.
RESULTS:
MBP enhanced the viability of U937 and Jurkat cells, and the effects were blocked by anti-TLR2, but not anti-TLR4 in U937 cells. Further studies confirmed that MBP was able to directly bind to U937 and Jurkat cells and modulate TLR expression. The effects of MBP depended on the activation of NF-κB and MAP kinase in U937 and Jurkat cells.
CONCLUSIONS:
Our results demonstrated that MBP could directly promote U937 cell viability via TLR2. It suggested that MBP may be used as an adjuvant for participating in the immunotherapy of haematologic malignancies.
OBJECTIVE AND DESIGN:
The anti-inflammatory effect of methyl-1-hydroxy-2-naphthoate (MHNA), a novel naphthol derivative, was evaluated in the lipopolysaccharide (LPS)-induced inflammatory response in murine macrophages.
MATERIALS AND METHODS:
The release of nitric oxide (NO), interleukin-1beta (IL-1β) and interleukin-6 (IL-6) were detected by the Griess reagent and ELISA methods. The protein expressions of inducible NO synthase (iNOS) and cyclooxygenase-2 (COX-2) were examined by Western blotting. The mRNA expressions of IL-1β, IL-6, iNOS and COX-2 were determined by real-time PCR. Activation of mitogen-activated protein kinases (MAPKs) and nuclear factor kappa B (NF-κB) pathways were detected by Western blotting, reporter gene assay and electrophoretic mobility shift assay.
RESULTS:
MHNA significantly inhibited the release of NO, IL-1β and IL-6 as well as the protein expression of iNOS and COX-2 in LPS-stimulated macrophages. It also inhibited the mRNA expression of iNOS, COX-2, IL-1β and IL-6. Further studies indicated that MHNA inhibited LPS-induced increases in NF-κB DNA-binding activity and NF-κB transcriptional activity as well as IκB-α degradation and NF-κB translocation in a dose-dependent manner. Meanwhile, the activation of p38 MAPK and c-Jun N-terminal kinases (JNK) induced by LPS were decreased by MHNA.
CONCLUSIONS:
MHNA inhibits the LPS-induced inflammatory response in murine macrophages via suppression of NF-κB and MAPKs signaling pathways activation.
PURPOSE:
This study was to investigate the clinicopathologic significance and potential role of HOXB7 in the development and progression of colorectal cancer (CRC).
EXPERIMENTAL DESIGN:
The relationship between HOXB7 expression and clinical characteristics of CRC was analyzed in 224 paraffin-embedded archived CRC specimens by immunohistochemistry (IHC). The effects of HOXB7 on cell growth and proliferation, as well as on tumorigenesis, were examined both in vitro and in vivo, using MTT assay, colony formation assay, cell cycle analysis, soft agar assay, and tumorigenesis in nude mice. Western blotting and real-time reverse transcriptase-PCR were performed to examine the impact of HOXB7 on the PI3K/Akt and MAPK signaling pathways.
RESULTS:
HOXB7 protein level was significantly correlated with advanced Dukes stage (P < 0.001), T stage (P = 0.012), distant metastasis (P = 0.042), higher proliferation index (P = 0.007) and poor survival of patients (P = 0.005). Enforced expression of HOXB7 in CRC cell lines significantly enhanced cell growth, proliferation and tumorigenesis. Conversely, knockdown of HOXB7 caused an inhibition of cell growth, proliferation, and tumorigenesis. We also showed that HOXB7 accelerated G(0)-G(1) to S-phase transition concomitantly with upregulation of cyclin D1 and downregulation of p27Kip1. On the contrary, knockdown of HOXB7 caused G(1)-S-phase arrest, downregulation of cyclin D1 and upregulation of p27Kip1. Enforced expression of HOXB7 could enhance PI3K/AKT and MAPK pathway activity.
CONCLUSION:
Our findings suggest that HOXB7 protein, as a valuable marker of CRC prognosis, plays an important role in the development and progression of human CRC.
©2011 AACR.
BACKGROUND:
Previous studies showed that connective tissue growth factor (CTGF)-induced proliferation of lung fibroblasts and production of chemokines in mesangial cells could be inhibited by lipoxin A(4) (LXA(4)). It is speculated that LXA(4) could modulate the CTGF-induced epithelial to mesenchymal transition.
METHODS:
The expressions of alpha-smooth muscle actin (alpha-SMA), E-cadherin, integrin-linked kinase (ILK), extracellular signal-regulated kinase 1/2 (ERK1/2), phosphatidylinositol 3-kinase (PI3-K), Akt and Smad signaling were assessed by Western blot and/or real-time RT-PCR, and activation of Ras or ILK by activity assay, expressions of alpha-SMA and zonula occludens-1 by immunofluorescence assay in proximal tubular epithelial cells (HK-2).
RESULTS:
Pretreatment of HK-2 cells with LXA(4) inhibited the morphological fibroblast-like changes and alpha-SMA expression induced by CTGF but not by transforming growth factor-beta(1) (TGF-beta(1)). The expressions of E-cadherin and zonula occludens-1 reduced by CTGF but not by TGF-beta(1) were increased by LXA(4). LXA(4) inhibited the expression and activity of ILK and activation of Ras, ERK1/2, PI3-K and Akt in HK-2 cells stimulated by CTGF. LXA(4) did not affect TGF-beta(1)-induced expression of ILK, Smad-2/3 phosphorylation and Smad-2's binding to Smad-4 and subsequent nuclear translocation.
CONCLUSION:
LXA(4) inhibits the tubular epithelial to mesenchymal transition, initiated by CTGF but not by TGF-beta(1), via downregulation of ILK, Ras/MEK/ERK1/2 and PI3-K/Akt-dependent signal pathway stimulated by CTGF.
2010 S. Karger AG, Basel.
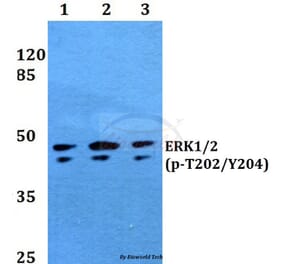

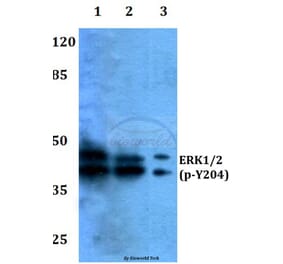
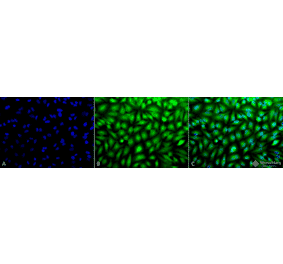
![Western Blot - Anti-ERK1 Antibody [ARC2591] (A306776) - Antibodies.com](https://cdn.antibodies.com/image/catalog/306/A306776_1.jpg?profile=product_alternative)