Unconjugated
Improved mouse models for type 1 diabetes (T1D) therapy development are needed. T1D susceptibility is restored to normally resistant NOD.ß2m-/- mice transgenically expressing human disease-associated HLA-A*02:01 or HLA-B*39:06 class I molecules in place of their murine counterparts. T1D is dependent on pathogenic CD8+ T-cell responses mediated by these human class I variants. NOD.ß2m-/--A2.1 mice were previously used to identify ß-cell autoantigens presented by this human class I variant to pathogenic CD8+ T cells and for testing therapies to attenuate such effectors. However, NOD.ß2m-/- mice also lack nonclassical MHC I family members, including FcRn, required for antigen presentation, and maintenance of serum IgG and albumin, precluding therapies dependent on these molecules. Hence, we used CRISPR/Cas9 to directly ablate the NOD H2-Kd and H2-Db classical class I variants either individually or in tandem (cMHCI-/-). Ablation of the H2-Ag7 class II variant in the latter stock created NOD mice totally lacking in classical murine MHC expression (cMHCI/II-/-). NOD-cMHCI-/- mice retained nonclassical MHC I molecule expression and FcRn activity. Transgenic expression of HLA-A2 or -B39 restored pathogenic CD8+ T-cell development and T1D susceptibility to NOD-cMHCI-/- mice. These next-generation HLA-humanized NOD models may provide improved platforms for T1D therapy development.
While a third of the world carries the burden of tuberculosis, disease control has been hindered by a lack of tools, including a rapid, point-of-care diagnostic and a protective vaccine. In many infectious diseases, antibodies (Abs) are powerful biomarkers and important immune mediators. However, in Mycobacterium tuberculosis (Mtb) infection, a discriminatory or protective role for humoral immunity remains unclear. Using an unbiased antibody profiling approach, we show that individuals with latent tuberculosis infection (Ltb) and active tuberculosis disease (Atb) have distinct Mtb-specific humoral responses, such that Ltb infection is associated with unique Ab Fc functional profiles, selective binding to Fc?RIII, and distinct Ab glycosylation patterns. Moreover, compared to Abs from Atb, Abs from Ltb drove enhanced phagolysosomal maturation, inflammasome activation, and, most importantly, macrophage killing of intracellular Mtb. Combined, these data point to a potential role for Fc-mediated Ab effector functions, tuned via differential glycosylation, in Mtb control.









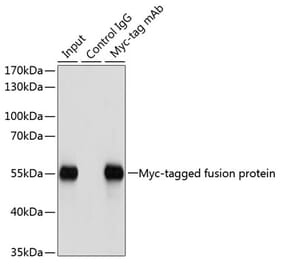
![Western Blot - Anti-Mouse IgG Antibody [RM104] (A121355) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121355_1.jpg?profile=product_alternative)

![ELISA - Anti-Mouse IgG Antibody [RMG07] (A121329) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121329_1.png?profile=product_alternative)
![Flow Cytometry - Rabbit IgG [ARC5105-03] - Isotype Control (A310007) - Antibodies.com](https://cdn.antibodies.com/image/catalog/310/A310007_1.jpg?profile=product_alternative)
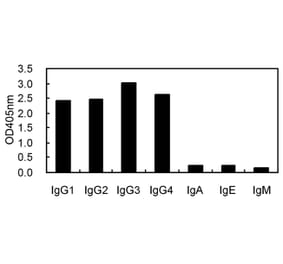
![ELISA - Anti-Rabbit IgG Antibody [RMG03] (A121342) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121342_1.png?profile=product_alternative)
![Western Blot - Anti-Mouse IgG Antibody [RM106] (A121356) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121356_1.jpg?profile=product_alternative)