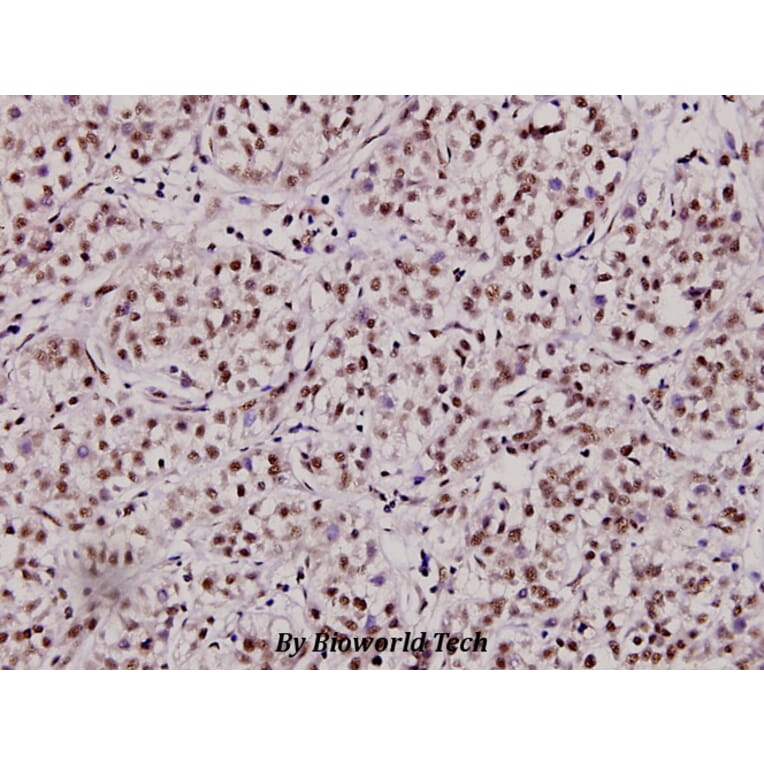
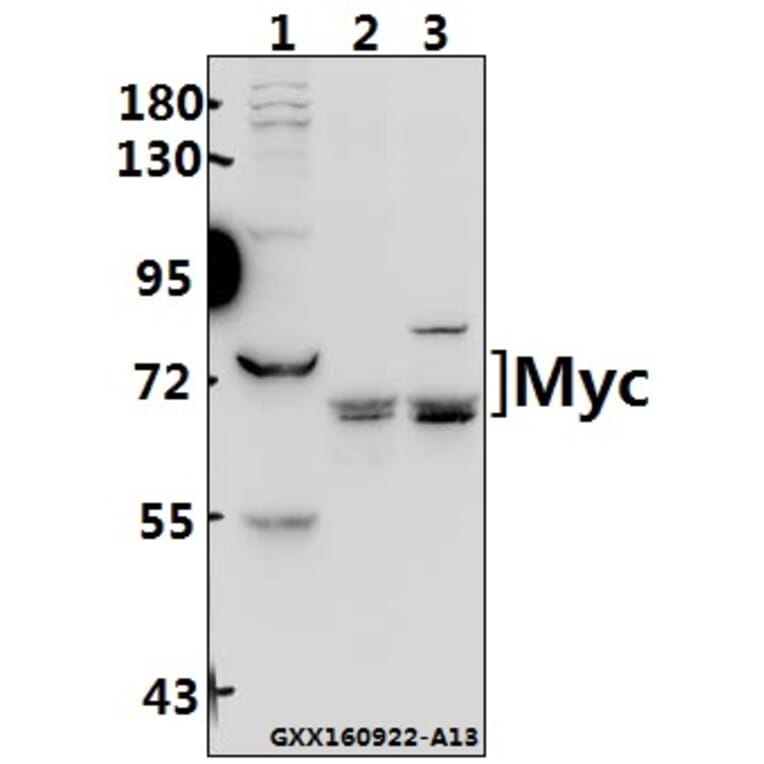
Unconjugated
Angiogenesis is associated with the progression of multiple myeloma (MM). Wogonin is an active mono-flavonoid with remarkable antitumor activity. However, its impact on MM-stimulated angiogenesis remains largely unknown. Here, we demonstrated that wogonin decreased expression and secretion of pro-angiogenic factors in MM cells via c-Myc/HIF-1α signaling axis, reducing MM-stimulated angiogenesis and MM cell proliferation in vivo. Overexpression of c-Myc in MM cells disrupted the balance between VHL SUMOylation and ubiquitination, and thus inhibited proteasome-mediated HIF-1α degradation. Impaired function of VHL ubiquitination complex in c-Myc-overexpressing cells was fully reversed by wogonin treatment via increasing HIF-1α-VHL interaction and promoting HIF-1α degradation. Collectively, our in vitro and in vivo studies reveal for the first time that wogonin represses MM-stimulated angiogenesis and tumor progression via c-Myc/VHL/HIF-1α signaling axis.
The purpose of this study was to elucidate the molecular mechanisms of microRNA-205 (miR-205) as a tumor suppressor in prostate cancer (PCa). In the present study, microRNA microarray analysis suggested that the expression of miR-205 was significantly decreased in advanced PCa compared with early PCa. Real-time PCR analysis also indicated that miR-205 expression was significantly decreased in PCa tissues compared with non-cancerous tissues. Moreover, the expression of miR-205 has been demonstrated to be associated with the clinicopathological stage and total/free prostate-specific antigen (PSA) level of PCa. Functional analyses showed that both the overexpression of miR-205 and the knockdown of c-SRC in PCa cell lines could inhibit cell growth, colony formation, migration, invasion and the cell cycle as well as induce cell apoptosis in vitro. Furthermore, over-expressing miR-205 reduced tumorigenicity in vivo. Through a luciferase activity assay and Western blotting, c-SRC was identified as a target of miR-205 in cells. The overexpression of miR-205 suppressed c-SRC and its downstream signaling molecules, including FAK, p-FAK, ERK1/2 and p-ERK1/2, and attenuated cell proliferation, invasion and tumor growth.
Aberrant promoter region hypermethylation of upstream transcription factors may be responsible for silencing entire anti-neoplastic gene networks. In this study, we explored whether transcription factor coding gene, caudal-related homeobox 2 (CDX2), is silenced by promoter hypermethylation in lung cancer, and examined its potential tumor-suppressive functions. Semi-quantitative RT-PCR showed that four of six lung cancer cell lines exhibited no or weak CDX2 expression. Expression of CDX2 was correlated to CDX2 promoter region methylation status, as determined by methylation-specific PCR (MSP) and bisulfite sequencing. Restoration of CDX2 expression was induced by treatment with demethylating drug 5-aza-2'-deoxycytidine (5-AZA) in lung cancer cell lines. Methylation of CDX2 was common in human primary lung cancer (61 of 110 tumors, 55.45%), but no methylation was found in normal lung tissues. Re-expression of CDX2 suppressed lung cancer cell proliferation and blocked cells in G1 phase. β-catenin/TCF activity and downstream genes expression were inhibited by re-expression of CDX2, and increased by depletion of CDX2. In conclusion, CDX2 is frequently methylated in lung cancer, and expression of CDX2 is regulated by promoter region hypermethylation. CDX2 may serve as a tumor suppressor in lung cancer and inhibits lung cancer cell proliferation by suppressing Wnt signaling.
Dapper, Dishevelled-associated antagonist of β-catenin (DACT), is involved in Xenopus embryonic development. Human DACT2 is localized on chromosome 6q27, a region of frequent loss of heterozygosity (LOH) in human cancers. However, the function and regulation of DACT2 in human lung cancer remain unclear. DNA sequencing, methylation-specific PCR (MSP), semi-quantitative RT-PCR, western blotting, and xenograft models were employed in this study. Eight lung cancer cell lines, 106 cases of primary lung cancer, four specimens of normal lung from patients without cancer, and 99 blood samples from healthy individuals were examined. We found that while there was no SNP related to lung cancer, the DACT2 promoter region is frequently methylated in human lung cancer. DACT2 is silenced by promoter region hypermethylation and re-expressed by 5-aza-2'-deoxyazacytidine treatment of lung cancer cell lines. Methylation of DACT2 was associated with poor differentiation of lung cancer. Loss of DACT2 expression was associated with promoter region hypermethylation in primary lung cancer, and was associated with increased β-catenin expression. Restoration of DACT2 expression suppressed tumour proliferation both in vitro and in vivo. DACT2 expression was down-regulated by siRNA knockdown in H727 cells. DACT2 inhibited T-cell factor/lymphoid enhancer factor (TCF/LEF) and its downstream genes. In conclusion, DACT2 methylation is a potential lung cancer detection marker. DACT2 is regulated by promoter region hypermethylation. DACT2 inhibits lung cancer proliferation by suppressing the Wnt signalling pathway in lung cancer.
Our previous studies indicated that JWA plays an important role in DNA damage repair, cell migration, and regulation of MAPKs. In this study, we investigated the role of JWA in chemical carcinogenesis using conditional JWA knockout (JWA(Δ2/Δ2)) mice and two-stage model of skin carcinogenesis. Our results indicated that JWA(Δ2/Δ2) mice were resistant to the development of skin papillomas initiated by 7, 12-dimethylbenz(a)anthracene (DMBA) followed by promotion with 12-O-tetradecanoylphorbol-13-acetate (TPA). In JWA(Δ2/Δ2) mice, the induction of papilloma was delayed, and the tumor number and size were reduced. In primary keratinocytes from JWA(Δ2/Δ2) mice, DMBA exposure induced more intensive DNA damage, while TPA-promoted cell proliferation was reduced. The further mechanistic studies showed that JWA deficiency blocked TPA-induced activation of MAPKs and its downstream transcription factor Elk1 both in vitro and in vivo. JWA(Δ2/Δ2) mice are resistance to tumorigenesis induced by DMBA/TPA probably through inhibition of transcription factor Elk1 via MAPKs. These results highlight the importance of JWA in skin homeostasis and in the process of skin tumor development.
BACKGROUND:
Natural products represent an important source for agents of cancer prevention and cancer treatment. More than 60% of conventional anticancer drugs are derived from natural sources, particularly from plant-derived materials. In this study, 2α, 3α, 19β, 23β-tetrahydroxyurs-12-en-28-oic acid (THA), a novel triterpenoid from the leaves of Sinojackia sarcocarpa, was isolated, and its anticancer activity was investigated both in vitro and in vivo.
PRINCIPAL FINDINGS:
THA possessed potent tumor selected toxicity in vitro. It exhibited significantly higher cytotoxicity to the cancer cell lines A2780 and HepG2 than to IOSE144 and QSG7701, two noncancerous cell lines derived from ovary epithelium and liver, respectively. Moreover, THA showed a dose-dependent inhibitory effect on A2780 ovary tumor growth in vivo in nude mice. THA induced a dose-dependent apoptosis and G2/M cell cycle arrest in A2780 and HepG2 cells. The THA-induced cell cycle arrest was accompanied by a downregulation of Cdc2. The apoptosis induced by THA was evident by induction of DNA fragmentation, release of cytoplasmic Cytochrome c from mitochondria, activation of caspases, downregulation of Bcl-2 and upregulation of Bax.
CONCLUSION:
The primary data indicated that THA exhibit a high toxicity toward two cancer cells than their respective non-cancerous counterparts and has a significant anticancer activity both in vitro and in vivo. Thus, THA and/or its derivatives may have great potential in the prevention and treatment of human ovary tumors and other malignancies.
Surgery-induced inflammation has been associated with cancer recurrence and metastasis in colorectal cancer (CRC). As a constituent of gram-negative bacteria, lipopolysaccharide (LPS) is frequently abundant in the peri-operative window. However, the definite roles of LPS in tumour progression remain elusive. Here we reported that LPS treatment increased PKM expression through activation of NF-κB signalling pathway, and knockdown of PKM reversed LPS-induced TNF-α, IL-1β production and cell proliferation in CRC cells. We further showed that the PKM2 but not PKM1 mediated the pro-inflammatory and proliferative effects of LPS. Interestingly, LPS promoted PKM2 binding to the STAT3 promoter to enhance STAT3 expression and its subsequent nuclear translocation. Depletion of STAT3 decreased PKM2-induced TNF-α and IL-1β expression, indicating that STAT3 mediates the pro-inflammatory effects of PKM2. Furthermore, it is the protein kinase activity but not the pyruvate kinase activity of PKM2 that is required for inflammatory cytokine production. Collectively, our findings reveal the NF-κB-PKM2-STAT3 axis as a novel mechanism for the regulation of TNF-α and IL-1β production and suggest the importance of PKM2 as a key inflammatory mediator in inflammatory microenvironment.
The metabolic activity in cancer cells primarily rely on aerobic glycolysis. Besides glycolysis, some tumor cells also exhibit excessive addition to glutamine, which constitutes an advantage for tumor growth. M2-type pyruvate kinase (PKM2) plays a pivotal role in sustaining aerobic glycolysis, pentose phosphate pathway and serine synthesis pathway. However, the participation of PKM2 in glutaminolysis is little to be known. Here we demonstrated that PKM2 depletion could provoke glutamine metabolism by enhancing the β-catenin signaling pathway and consequently promoting its downstream c-Myc-mediated glutamine metabolism in colon cancer cells. Treatment with 2-deoxy-d-glucose (2-DG), a glycolytic inhibitor, got consistent results with the above. In addition, the dimeric form of PKM2, which lacks the pyruvate kinase activities, plays a critical role in regulating β-catenin. Moreover, we found that overexpression of PKM2 negatively regulated β-catenin through miR-200a. These insights supply evidence that glutaminolysis plays a compensatory role for cell survival upon glucose metabolism impaired.
Dichlorodiphenyltrichloroethane (DDT) is a persistent organic pollutant, involved in the progression of many cancers, including liver cancer. However, the underlying mechanism(s) of DDT, especially how low doses DDT cause liver cancer, is poorly understood. In this study, we evaluated the impact of p,p'-DDT on the growth of hepatocellular carcinoma using both in vitro and in vivo models. The present data indicated that the proliferation of HepG2 cells was strikingly promoted after exposed to p,p'-DDT for 4 days. In addition, reactive oxygen species (ROS) content was significantly elevated, accompanied with inhibitions of γ-glutamylcysteine synthetase (γ-GCS) and superoxide dismutase (SOD) activities. Interestingly, the levels of β-catenin and its downstream target genes (c-Myc and CyclinD1) were significantly up-regulated, and co-treatment of NAC, the ROS inhibitor, inhibited these over-expressed proteins. Moreover, the p,p'-DDT-stimulated proliferation of HepG2 cells could be reversed after NAC or β-catenin siRNA co-treatment. Likewise, p,p'-DDT treatment increased the growth of tumor in nude mice, stimulated oxidative stress and Wnt/β-catenin pathway. Our study indicates that low doses p,p'-DDT exposure promote the growth of hepatocellular carcinoma via Wnt/β-catenin pathway which is activated by oxidative stress. The finding suggests an association between low dose DDT exposure and liver cancer growth.
Rare genetic mutations in the DJ-1 and Parkin genes cause recessive Parkinsonism, however, the relationship between these two genes is not fully elucidated. Current emerging evidence suggests that these genes are involved in mitochondrial homeostasis, and that a deficiency in either of these two genes is associated with damages in mitochondrial function and morphology. In this study, we demonstrated that knockdown of DJ-1 expression or the overexpression of the DJ-1 L166P mutation results in a damaged phenotype in mitochondria and a hypersensitivity to H2O2-induced cell apoptosis. These phenotypes result from increased levels of endogenous oxidative stress. However, overexpression of wild-type Parkin rescued the phenotypes observed in the mitochondria of DJ-1 knockdown and DJ-1 L166P mutant cells. We also determined that there were differences between the two cell models. Furthermore, both H₂O₂ treatment and the DJ-1 L166P mutation weakened the interaction between DJ-1 and Parkin. Taken together, these findings suggested that DJ-1 and Parkin were linked through oxidative stress, and that overexpression of Parkin protects DJ-1 protein-deficient and DJ-1 L166P mutant-expressing cells via inhibition of oxidative stress.
The RNA-binding protein Musashi2 (Msi2) has been identified as a master regulator within a variety of stem cell populations via the regulation of translational gene expression. A recent study has suggested that Msi2 is strongly expressed in leukemic cells of acute myeloid leukemia patients, and elevated Msi2 is associated with poor prognosis. However, the potential role of Msi2 in leukemogenesis is still not well understood. Here, we investigated the effect of Msi2 knockdown on the biological properties of leukemic cells. High expression of Msi2 was found in K562 and KG-1a leukemic cell lines, and low expression was observed in the U937 cell line. We transduced K562 cells with two independent adenoviral shRNA vectors targeting Msi2 and confirmed knockdown of Msi2 at the mRNA and protein levels. Msi2 silencing inhibited cell growth and caused cell cycle arrest by increasing the expression of p21 and decreasing the expression of cyclin D1 and cdk2. In addition, knockdown of Msi2 promoted cellular apoptosis via the upregulation of Bax and downregulation of Bcl-2 expression. Furthermore, Msi2 knockdown resulted in the inactivation of the ERK/MAPK and p38/MAPK pathways, but no remarkable change in p-AKT was observed. These data provide evidence that Msi2 plays an important role in leukemogenesis involving the MAPK signaling pathway, which indicates that Msi2 may be a novel target for leukemia treatment.
Wogonin, a naturally occurring mono-flavonoid, has been reported to have tumor therapeutic potential and good selectivity both in vitro and in vivo. Herein, we investigated the anti-proliferation effects and associated mechanisms of wogonin in human colorectal cancer in vitro. The flow-cytometric analysis showed that wogonin induced a G1 phase cell cycle arrest in HCT116 cells in a concentration- and time-dependent manner. Meanwhile, the cell cycle-related proteins, such as cyclin A, E, D1, and CDK2, 4 were down-regulated in wogonin-induced G1 cell cycle arrest. Furthermore, we showed that the anti-proliferation and G1 arrest effect of wogonin on HCT116 cells was associated with deregulation of Wnt/β-catenin signaling pathway. Wogonin-treated cells showed decreased intracellular levels of Wnt proteins, and activated degradation complex to phosphorylated and targeted β-catenin for proteasomal degradation. Wogonin inhibited β-catenin-mediated transcription by interfering in the transcriptional activity of TCF/Lef, and repressing the kinase activity of CDK8 which has been considered as an oncogene involving in the development of colorectal cancers. Moreover, CDK8 siRNA-transfected HCT116 cells showed similar results to wogonin treated cells. Thus, our data suggested that wogonin induced anti-proliferation and G1 arrest via Wnt/β-catenin signaling pathway and it can be developed as a therapeutic agent against human colorectal cancer.
Cellular senescence is a growth-arrest program that limits cell proliferation. Low-power laser irradiation (LPLI) has been demonstrated to promote cell proliferation. However, whether LPLI can inhibit cellular senescence remains unknown. In the present study, to investigate the functional role of LPLI against skin aging, we used ultraviolet radiation b (UVB) to induce cell senescence. We first report that LPLI can delay UVB-induced cell senescence. The senescence-associated β-galactosidase (SA-β-Gal) activity and p21 expression, hallmarks of senescent cells, were decreased in the Forkhead box transcription factor FOXM1-dependent manner under treatment with LPLI. The effect of LPLI was further enhanced with an overexpression of FOXM1, and abolished when FOXM1 was knockdown with short hairpin RNA (shRNA). Furthermore, LPLI activated the extracellular regulated protein kinases (ERK) that was upstream of FOXM1. This led to FOXM1 phosphorylation and nuclear translocation. Nuclear translocation enhanced FOXM1 transcriptional activity and promoted its downstream target gene c-Myc expression that could inhibit p21 expression. These findings highlight the protective effects of ERK/FOXM1 pathway against UVB-induced cell senescence, suggesting a potential protecting strategy for treating skin aging by LPLI.
Interferon regulatory factor (IRF) 3, a member of the highly conserved IRF family transcription factors, plays a pivotal role in innate immune response, apoptosis, and oncogenesis. Recent studies have implicated IRF3 in a wide range of host defense. However, whether IRF3 induces defensive responses to hypertrophic stresses such as biomechanical stress and neurohumoral factors remains unclear. Herein, we employed an IRF3-deficient mouse model, cardiac-specific IRF3-overexpression mouse model and isolated cardiomyocytes to investigate the role of IRF3 in cardiac hypertrophy induced by aortic banding (AB) or isoproterenol (ISO). The extent of cardiac hypertrophy was quantitated by echocardiography as well as by pathological and molecular analysis. Our results demonstrate that IRF3 deficiency profoundly exacerbated cardiac hypertrophy, whereas overexpression of IRF3 in the heart significantly blunted pathological cardiac remodeling induced by pressure overload. Similar results were also observed in cultured cardiomyocytes upon the treatment with ISO. Mechanistically, we discovered that IRF3 interacted with ERK2 and thereby inhibited the ERK1/2 signaling. Furthermore, inactivation of ERK1/2 by U0126 offset the IRF3-deficient-mediated hypertrophic response induced by aortic banding. Altogether, these data demonstrate that IRF3 plays a protective role in AB-induced hypertrophic response by inactivating ERK1/2 in the heart. Therefore, IRF3 could be a new target for the prevention and therapy of cardiac hypertrophy and failure.
Emerging evidence indicates that the miR-17 family may have a causal role in human cancer tumorigenesis, but their specific effects on the occurrence of CRC (colorectal carcinoma) are still poorly understood. In the present study, we profiled CRC tissue samples by miRNA (microRNA) microarray and found that four members of the miR-17 family had higher expression in CRC tissues than in normal tissues. This finding was further validated by qRT-PCR (quantitative reverse transcription PCR). Transfecting CRC cells with an inhibitor of miR-17 lowered their ability to proliferate and induced G0/G1 arrest. We also confirmed that miR-17 exerted this function by directly targeting RND3 in vitro, and that the expression of miR-17 was negatively correlated with that of RND3 in CRC tissues and CRC cells. Moreover, miR-17 inhibition led to tumour growth suppression and up-regulation of RND3 expression in a nude mouse xenograft model. RND3 expression was found to be significantly lower in CRC tissues than in normal tissues and adenomas, indicating that RND3 may act as a tumour suppressor gene in CRC. In conclusion, the present study suggests that miR-17 plays an important role in CRC carcinogenesis by targeting RND3 and may be a therapeutic agent for CRC.
Ursolic acid (UA), a pentacyclic triterpenoid compound, is widely distributed in the plant kingdom and has a broad range of biological effects. This study was carried out for the first time to investigate the potential role of UA in the differentiation of human leukemia HL60 cells and the underlying mechanisms in it. UA could induce differentiation of HL60 cells into the monocytic lineage, as assessed by the morphological change, nitroblue tetrazolium reduction assay, and expression of CD14 and CD11b surface antigens. Moreover, UA activated the extracellular signal-regulated kinase (ERK) pathway in both dose-dependent and time-dependent manners. Inhibiting ERK pathway activation with PD98059 could significantly block the differentiation induced by UA. Consistent with the induced differentiation, the upregulation of CCAAT/enhancer-binding protein β by UA was also eliminated by PD98059. Taken together, the results reported here show that UA can promote the monocytic differentiation of HL60 cells and increase the expression of CCAAT/enhancer-binding protein β by activating the ERK pathway, suggesting that UA could be a potential candidate as a differentiation-inducing agent for the therapeutic treatment of leukemia.
AIM:
To examine epigenetic changes and the function of HOXA11 in human gastric cancer (GC).
MATERIALS & METHODS:
Seven GC cell lines, five cases of normal gastric mucosa and 112 cases primary GC samples were used in this study.
RESULTS:
Expression of HOXA11 and lack of promoter region methylation were found in NCI-N87, MKN45, BGC823 and HGC27 cells. Loss of expression and complete methylation were found in AGS gastric cancer cells. Reduced expression and partial methylation were found in MGC803 and SGC7901 cells. Restoration of HOXA11 expression was induced by 5-aza-2'-deoxycytidine. HOXA11 was methylated in 81.25% (91/112) of primary GCs. The presence of methylation was associated with male gender, tumor size, tumor differentiation and lymph node metastasis (all p < 0.05). Restoration of HOXA11 expression reduced cell proliferation, invasion, migration and induced apoptosis and G2/M phase arrest. HOXA11 was found to inhibit Wnt signaling by upregulating NKD1 expression.
CONCLUSION:
Epigenetic silencing of HOXA11 promotes GC proliferation, migration and invasion through activation of Wnt signaling.
BACKGROUND:
Proliferation of vascular smooth muscle cells (VSMCs) is a crucial event in the pathogenesis of intimal hyperplasia, which is the main cause of restenosis after vascular reconstruction. In this study, we assessed the impact of let-7a microRNA (miRNA) on the proliferation of VSMCs.
METHODS:
Using miRNA microarrays analysis for miRNA expression in the vein graft model. Lentiviral vector-mediated let-7a was transfected into the vein grafts. In situ hybridization was performed to detect let-7a. Cultured rat VSMCs were transfected with let-7a mimics for different periods of time. Cell proliferation, migration and cell cycle activity were monitored following transfection of the let-7a mimics. Immunohistochemical and Western blotting analysis the expression levels of c-myc and K-ras.
RESULTS:
We found that let-7a was the most downregulated miRNA in the vein graft model. In vivo proliferation of VSMCs was assessed in a rat model of venous graft intimal hyperplasia. Let-7a was found to localize mainly to the VSMCs. Let-7a miRNA expression was increased in VSMCs in the neointima of the let-7a treated group. Intimal hyperplasia was suppressed by upregulation of let-7a via lentiviral vector-mediated mimics. In cultured VSMCs, the expression of let-7a increased upon starving, and the upregulation of let-7a miRNA significantly decreased cell proliferation and migration. Immunohistochemical and Western blotting analysis demonstrated that treatment with let-7a mimics resulted in decreased expression levels of c-myc and K-ras.
CONCLUSIONS:
The results indicate that let-7a miRNA is a novel regulator of VSMC proliferation in intimal hyperplasia. These findings suggest that let-7a miRNA is a promising therapeutic target for the prevention of intimal hyperplasia.
Copyright © 2014 Elsevier Inc. All rights reserved.
OBJECTIVE:
To investigate the role of B-cell-specific Moloney murine leukemia virus integration site 1 (Bmi-1) in gastric cancer (GC) and its relationship with the clinicopathological features of GC.
METHODS:
Laser capture microdissection combined with real-time polymerase chain reaction and Western blot were used to determine the expressions of Bmi-1, the cellular homologue of avian myelocytomatosis virus (c-Myc), enhancer of Zeste homolog 2 (EZH2), phosphatase and tensin homologue (PTEN) and epithelial cadherin (E-cadherin) in 20 GC specimens and the adjacent non-cancerous gastric tissues.
RESULTS:
The mRNA and protein expressions of Bmi-1 in GC were increased compared with those of the non-cancerous gastric tissues (P = 0.012 and P = 0.000, respectively). Bmi-1 mRNA expression was positively correlated with tumor size, degree of tumor differentiation, invasion and lymph node metastasis. At both mRNA and protein levels, Bmi-1 was positively correlated with c-Myc and EZH2, but negatively correlated with PTEN and E-cadherin.
CONCLUSION:
Bmi-1 might be involved in GC at both transcription and translation levels.
© 2014 Chinese Medical Association Shanghai Branch, Chinese Society of Gastroenterology, Renji Hospital Affiliated to Shanghai Jiaotong University School of Medicine and Wiley Publishing Asia Pty Ltd.
Human dachshund homolog 1 (DACH1) is a major component of the Retinal Determination Gene Network (RDGN) and functions as a tumor suppressor. However, the regulation of DACH1 expression and its function in hepatocellular carcinoma (HCC) remain unclear. In this study, epigenetic changes of DACH1 were analyzed in HCC cell lines and primary cancers. We found that promoter region hypermethylation was correlated with loss or reduction of DACH1 expression, and restoration of DACH1 expression was induced by 5-aza-2'-deoxycytidine (5-AZA) in HCC cell lines. Promoter region methylation was found in 42% of primary HCC. Reduced expression of DACH1 was associated with poor differentiation of HCC nodules and higher serum aspartate aminotransferase/alanine aminotransferase ratio. DACH1 suppressed cellular growth by reactivating transforming growth factor beta (TGF-β) signaling. Ectopic expression of DACH1 enhanced chemosensitivity to 5-fluorouracil (5-FU) by inducing p21 expression in HCC cells.
CONCLUSION:
DACH1 is frequently methylated in HCC and DACH1 expression is regulated by promoter hypermethylation. Down-regulation of DACH1 is a novel mechanism for gaining resistance to the antiproliferative signaling of TGF-β1 and 5-FU resistance.
Copyright © 2013 by the American Association for the Study of Liver Diseases.
BACKGROUND:
Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) presents great challenges in the treatment of non-small cell lung cancer (NSCLC) patients, while the mechanisms are still not well understood. The β-catenin signaling pathway has been found to be associated with chemoresistance and can activate the EGFR and its downstream pathways. This study aimed to investigate the role of β-catenin in acquired resistance to EGFR-TKIs in NSCLC cell lines.
METHODS:
The expression and transcriptional activity of β-catenin were measured in both the NSCLC cell line PC9 and its sub-line PC9/AB(2) which has acquired resistance to gefitinib. Knockdown and overexpression of β-catenin in the PC9/AB(2) and PC9 cells were performed. The cell survival rate and the activation of the EGFR and its downstream pathways were detected in the two cell lines after transfection.
RESULTS:
Nuclear translocation of β-catenin was increased in the PC9/AB(2) cells and the baseline expression of members of the β-catenin signaling pathway was also higher in the PC9/AB(2) cells. Knocking down the expression of β-catenin increased the sensitivity of the PC9/AB(2) cells to gefitinib by blocking the activation of the EGFR downstream pathways, while β-catenin overexpression improved PC9 cells resistance to gefitinib by enhancing the activation of the EGFR and its downstream signaling.
CONCLUSION:
β-catenin plays an important role in acquired resistance to EGFR-TKIs in NSCLC cell lines and may be a potential therapeutic target for NSCLC patients who have failed to respond to targeted therapy.
Crown Copyright © 2013. Published by Elsevier Ltd. All rights reserved.
BACKGROUND/AIMS:
ω3-polyunsaturated fatty acids (ω3- PUFAs) are known to possess anticancer properties. However, the relationship between ω3-PUFAs and β-catenin, one of the key components of the Wnt signaling pathway, in human pancreatic cancer remains poorly characterized.
METHODS:
Human pancreatic cancer cells (SW1990 and PANC-1) were exposed to two ω3-PUFAs, docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), to investigate the relationship between ω3-PUFAs and the Wnt/β-catenin signaling pathway in vitro. Mouse pancreatic cancer (PANC02) cells were implanted into fat-1 transgenic mice, which express ω3 desaturases and result in elevated levels of ω3-PUFAs endogenously. The tumor size, levels of Wnt/β-catenin signaling molecules and apoptosis levels were analyzed to examine the influence of ω3-PUFAs in vivo.
RESULTS:
DHA and EPA significantly inhibited cell growth and increased cell death in pancreatic cancer cells. DHA also reduced β-catenin expression, T cell factor/lymphoid-enhancing factor reporter activity and induced β-catenin/Axin/GSK-3β complex formation, a known precursor to β-catenin degradation. Furthermore, Wnt3a, a natural canonical Wnt pathway ligand, reversed DHA-induced growth inhibition in PANC-1 cells. Immunohistochemical analysis showed aberrant upregulation and increased nuclear staining of β-catenin in tumor tissues from pancreatic cancer patients. However, β-catenin levels in tumor tissues from fat-1 transgenic mice were reduced with a significant increase in apoptosis compared with those from control mice.
CONCLUSION:
ω3-PUFAs may be an effective therapy for the chemoprevention and treatment of human pancreatic cancer. and IAP.
Copyright © 2011 S. Karger AG, Basel.