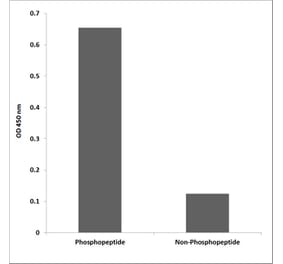
Unconjugated
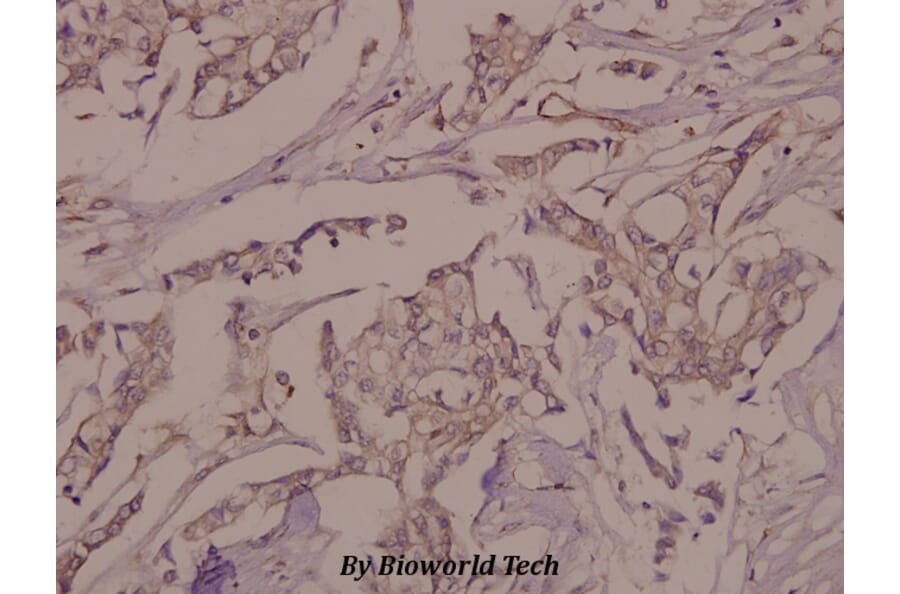
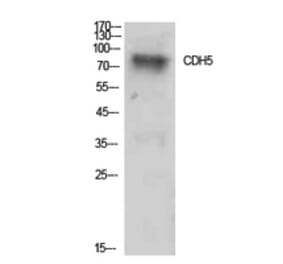
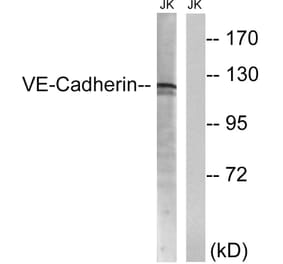
Wogonin, a naturally occurring monoflavonoid extracted from the root of Scutellaria baicalensis Georgi, has been shown to have anti-inflammatory and anti-tumor activities and inhibits oxidant stress-induced vascular permeability. However, the influence of wogonin on vascular hyperpermeability induced by overabounded inflammatory factors often appears in inflammatory diseases and tumor is not well known. In this study, we evaluate the effects of wogonin on LPS induced vascular permeability in human umbilical vein endothelial cells (HUVECs) and investigate the underlying mechanisms. We find that wogonin suppresses the LPS-stimulated hyperactivity and cytoskeleton remodeling of HUVECs, promotes the expression of junctional proteins including VE-Cadherin, Claudin-5 and ZO-1, as well as inhibits the invasion of MDA-MB-231 across EC monolayer. Miles vascular permeability assay proves that wogonin can restrain the extravasated Evans in vivo. The mechanism studies reveal that the expressions of TLR4, p-PLC, p-MLCK and p-MLC are decreased by wogonin without changing the total steady state protein levels of PLC, MLCK and MLC. Moreover, wogonin can also inhibit KCl-activated MLCK/MLC pathway, and further affect vascular permeability. Significantly, compared with wortmannin, the inhibitor of MLCK/MLC pathway, wogonin exhibits similar inhibition effects on the expression of p-MLCK, p-MLC and LPS-induced vascular hyperpermeability. Taken together, wogonin can inhibit LPS-induced vascular permeability by suppressing the MLCK/MLC pathway, suggesting a therapeutic potential for the diseases associated with the development of both inflammatory and tumor.
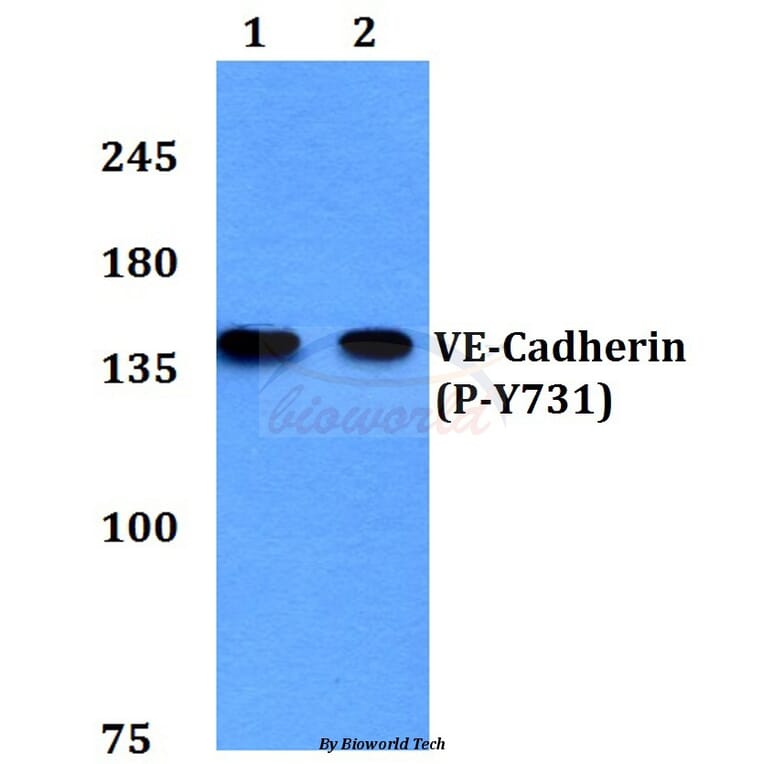
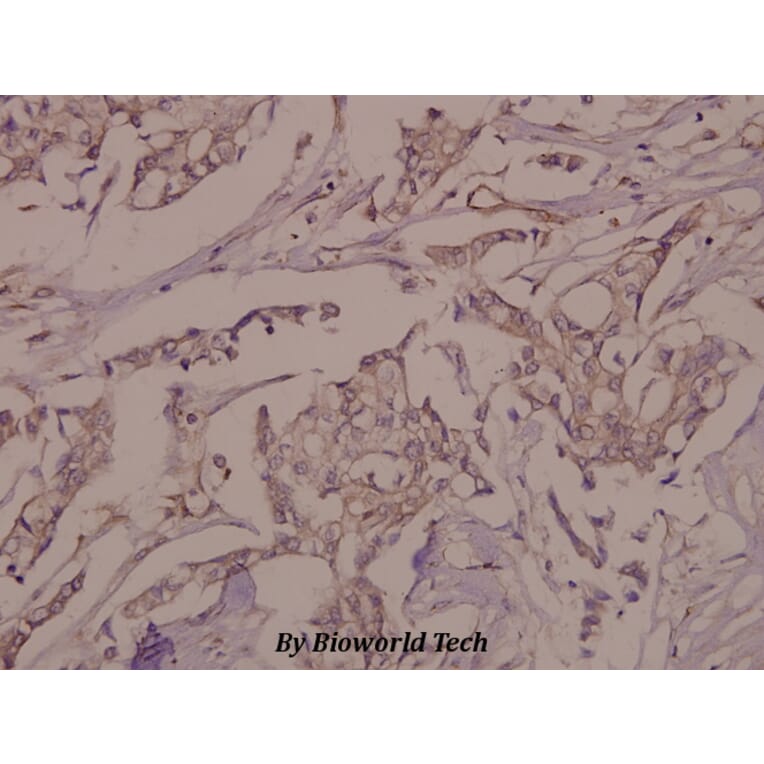
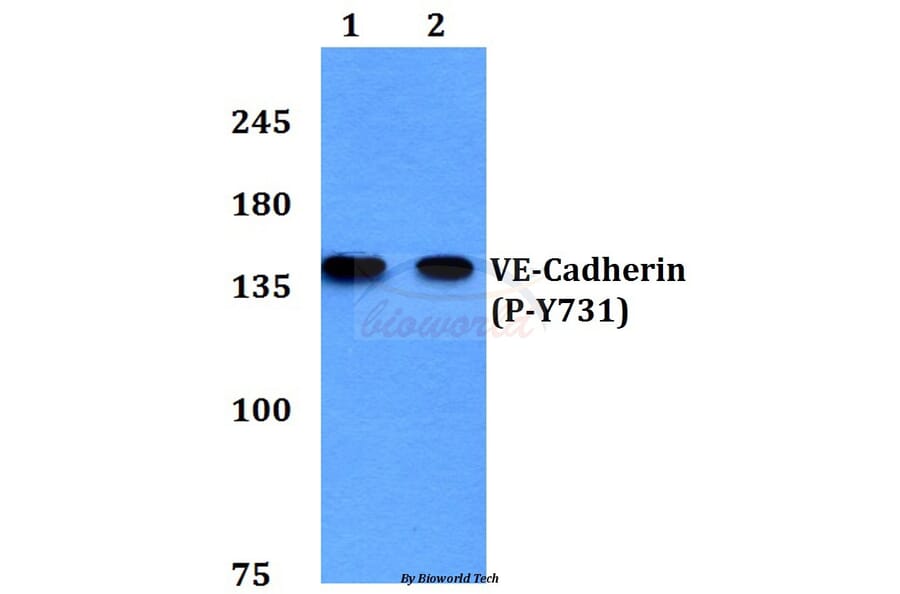
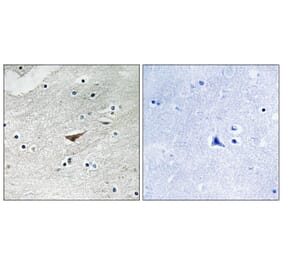
Tyrosine phosphorylation of the adhesion molecule VE-cadherin is assumed to affect endothelial junction integrity. However, it remains unclear whether tyrosine residues of VE-cadherin are required for the induction of vascular permeability and the regulation of leukocyte extravasation in vivo. We found here that knock-in mice expressing a Y685F mutant of VE-cadherin had impaired induction of vascular permeability, but those expressing a Y731F mutant did not. In contrast, mice expressing the Y731F VE-cadherin mutant showed decreased neutrophil-extravasation in cremaster tissue, but those expressing the Y685F mutant did not. Whereas inflammatory mediators induced the phosphorylation of Tyr685 in vivo, Tyr731 showed high baseline phosphorylation. Leukocytes triggered dephosphorylation of Tyr731 via the tyrosine phosphatase SHP-2, which allowed the adaptin AP-2 to bind and initiate endocytosis of VE-cadherin. Thus, Tyr685 and Tyr731 of VE-cadherin distinctly and selectively regulate the induction of vascular permeability or leukocyte extravasation.