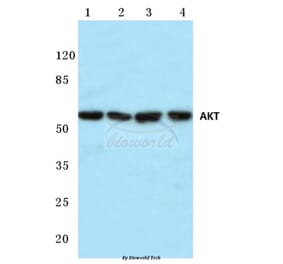
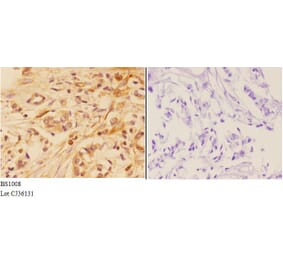
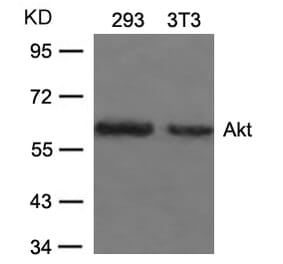
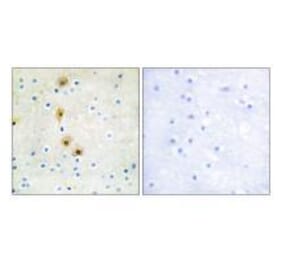
Unconjugated
BACKGROUND:
Fatty acid synthase (FASN) is frequently activated and overexpressed in human cancers, and plays a crucial role in the carcinogenesis of various cancers. In this study, our aims were to explore the role of FASN in regulating the "HER2-PI3K/Akt axis" activity and malignant phenotype of colorectal cancer.
METHODS:
Caco-2 cells with a high expression of both HER2 and FASN were selected for functional characterization. Caco-2 cells were transfected with either the FASN specific RNAi plasmid or the negative control RNAi plasmid, followed by the RT-qPCR and western blot to examine the expression of FASN, HER2, PI3K and Akt. The MTT and colony formation assays were used to assess the proliferation potential. The migration was investigated by the transwell, and the apoptosis and cell cycle were assayed by the flow cytometry.
RESULTS:
Notably, the expression of FASN, HER2, PI3K and Akt were downregulated upon a silence of FASN. The proliferation was decreased after a downregulation of FASN, which was consistent with an increased apoptosis rate. The migration was also impaired in FASN-silenced cells.
CONCLUSION:
A downregulation of FASN effectively inhibits the activity of "HER2-PI3K/Akt axis" and alters the malignant phenotype in colorectal cancer cells.
T-cadherin is an atypical member of the cadherin family, which lacks the transmembrane and intracellular domains and is attached to the plasma membrane via a glycosylphosphatidylinositol anchor. Unlike canonical cadherins, it is believed to function primarily as a signaling molecule. T-cadherin is highly expressed in endothelium. Using transendothelial electrical resistance measurements and siRNA-mediated depletion of T-cadherin in human umbilical vein endothelial cells, we examined its involvement in regulation of endothelial barrier. We found that in resting confluent monolayers adjusted either to 1% or 10% serum, T-cadherin depletion modestly, but consistently reduced transendothelial resistance. This was accompanied by increased phosphorylation of Akt and LIM kinase, reduced phosphorylation of p38 MAP kinase, but no difference in tubulin acetylation and in phosphorylation of an actin filament severing protein cofilin and myosin light chain kinase. Serum stimulation elicited a biphasic increase in resistance with peaks at 0.5 and 4-5 h, which was suppressed by a PI3 kinase/Akt inhibitor wortmannin and a p38 inhibitor SB 239063. T-cadherin depletion increased transendothelial resistance between the two peaks and reduced the amplitude of the second peak. T-cadherin depletion abrogated serum-induced Akt phosphorylation at Thr308 and reduced phosphorylation at Ser473, reduced phosphorylation of cofilin, and accelerated tubulin deacetylation. Adiponectin slightly improved transendothelial resistance irrespectively of T-cadherin depletion. T-cadherin depletion also resulted in a reduced sensitivity and delayed responses to thrombin. These data implicate T-cadherin in regulation of endothelial barrier function, and suggest a complex signaling network that links T-cadherin and regulation of barrier function.
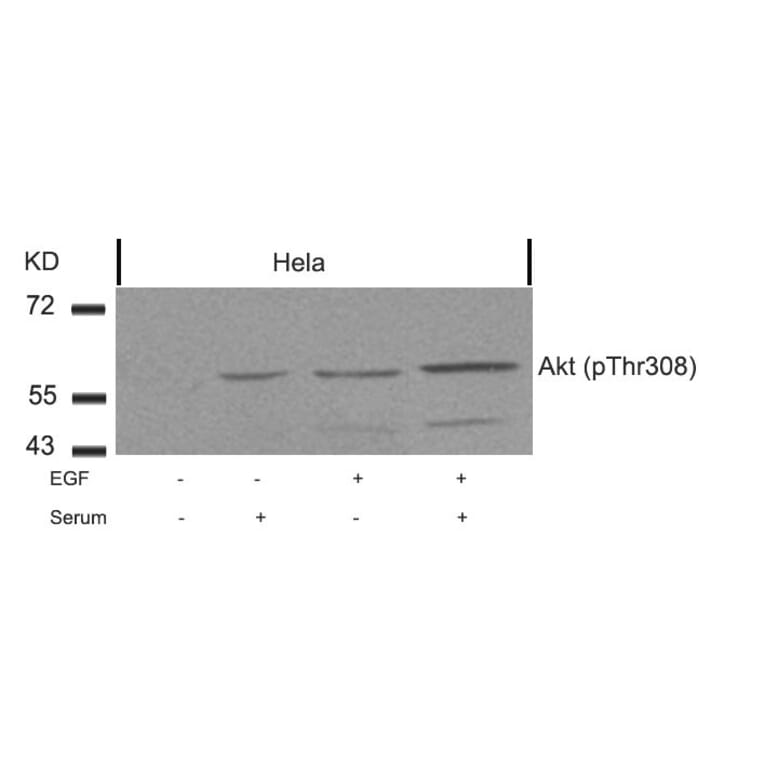
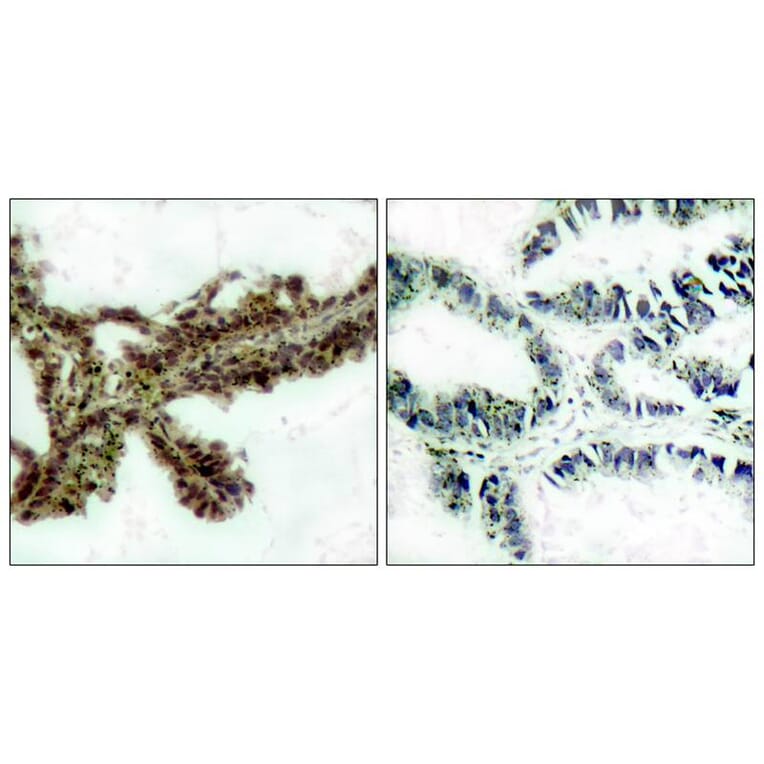
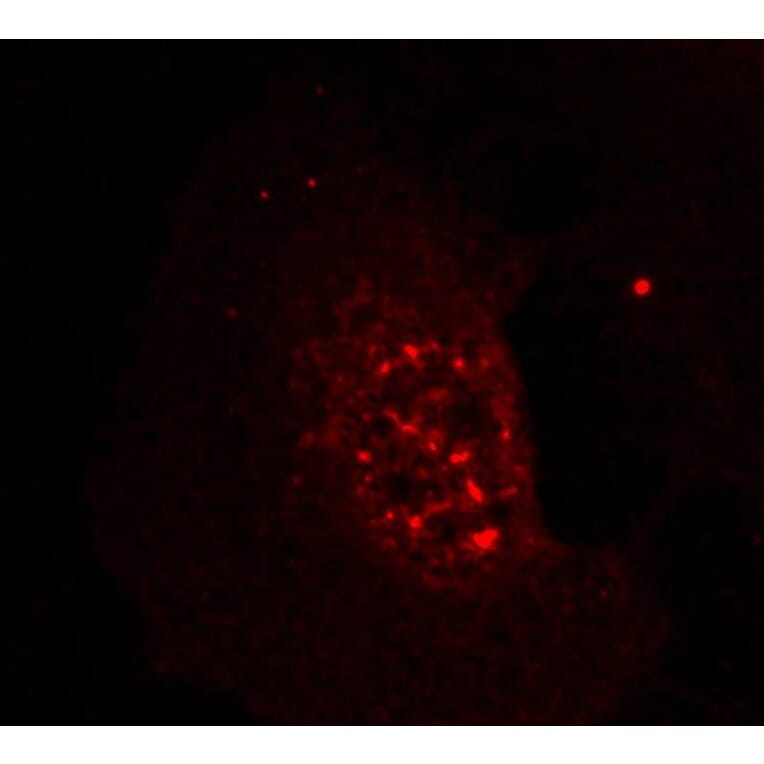
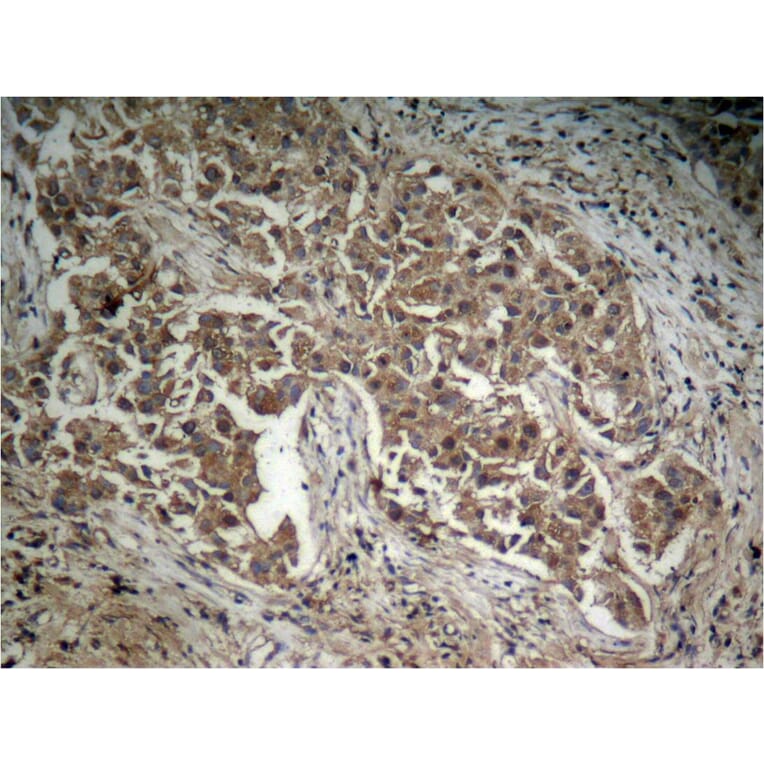
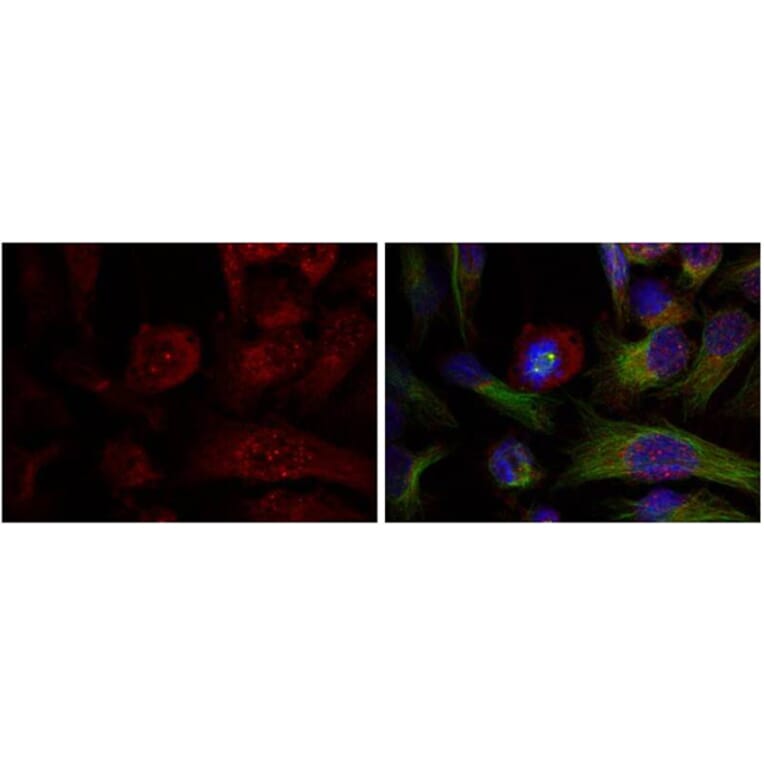





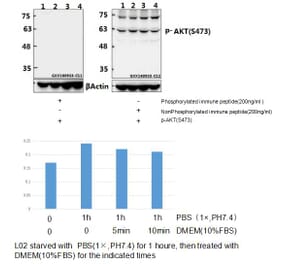
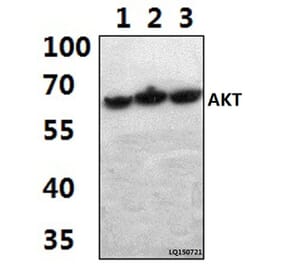
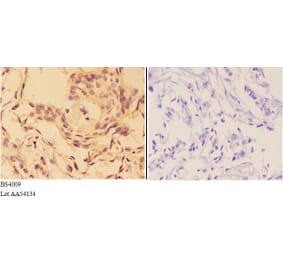
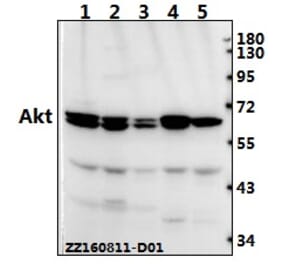
![Immunohistochemistry - Anti-AKT Antibody [RM316] (A121372) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121394_1.png?profile=product_alternative)
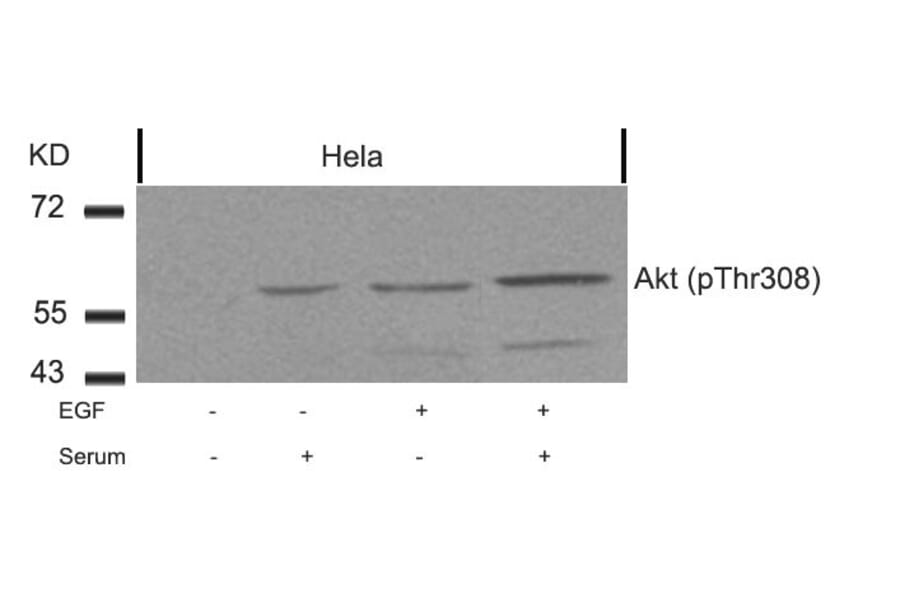
![Western Blot - Anti-AKT (phospho Ser473) Antibody [RM251] (A121248) - Antibodies.com](https://cdn.antibodies.com/image/catalog/121/A121248_1.png?profile=product_alternative)