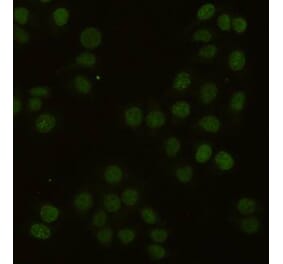
Immunocytochemistry staining of PCNA in PHA-stimulated lymphocytes using Anti-PCNA Antibody [PC10] (A86878), (diluted 1:400), detected with GAM IgG-Alexa Fluor®488 (diluted 1:200); DNA stained with propidium iodide. Colocalization of green PCNA signal and red DNA signal indicated as yellow.


![Flow Cytometry - Anti-PCNA Antibody [PC10] (A86878)](https://cdn.antibodies.com/image/catalog/86/A86878_1.jpg?profile=product_top)
![ICC - Anti-PCNA Antibody [PC10] (A86878)](https://cdn.antibodies.com/image/catalog/86/A86878_2.jpg?profile=product_top)
![IHC - Anti-PCNA Antibody [PC10] (A86878)](https://cdn.antibodies.com/image/catalog/86/A86878_3.jpg?profile=product_top)
![Flow Cytometry - Anti-PCNA Antibody [PC10] (A86878)](https://cdn.antibodies.com/image/catalog/86/A86878_1.jpg?profile=product_top_thumb)
![ICC - Anti-PCNA Antibody [PC10] (A86878)](https://cdn.antibodies.com/image/catalog/86/A86878_2.jpg?profile=product_top_thumb)
![IHC - Anti-PCNA Antibody [PC10] (A86878)](https://cdn.antibodies.com/image/catalog/86/A86878_3.jpg?profile=product_top_thumb)
![Flow Cytometry - Anti-PCNA Antibody [PC10] (A86878)](https://cdn.antibodies.com/image/catalog/86/A86878_1.jpg?profile=product_image)
![ICC - Anti-PCNA Antibody [PC10] (A86878)](https://cdn.antibodies.com/image/catalog/86/A86878_2.jpg?profile=product_image)
![IHC - Anti-PCNA Antibody [PC10] (A86878)](https://cdn.antibodies.com/image/catalog/86/A86878_3.jpg?profile=product_image)
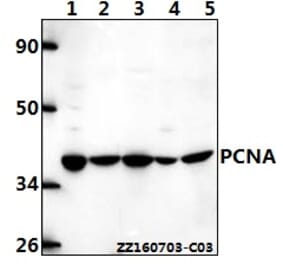
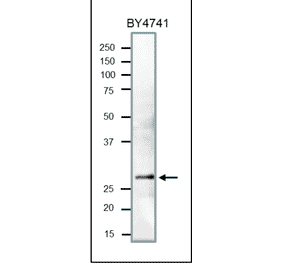
![SDS-PAGE - Anti-PCNA Antibody [PC10] (A281549) - Antibodies.com](https://cdn.antibodies.com/image/catalog/281/A281549_3.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-PCNA Antibody [PC10] (A252744) - Antibodies.com](https://cdn.antibodies.com/image/catalog/249/A249578_1.jpg?profile=product_alternative)
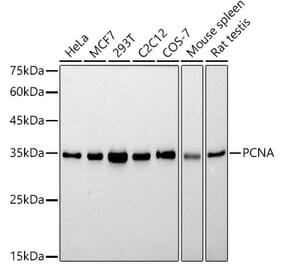
![Immunohistochemistry - Anti-PCNA Antibody [PC10] - BSA and Azide free (A249578) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252758_1.jpg?profile=product_alternative)
![Immunohistochemistry - Anti-PCNA Antibody [SPM350] (A252760) - Antibodies.com](https://cdn.antibodies.com/image/catalog/249/A249579_1.jpg?profile=product_alternative)
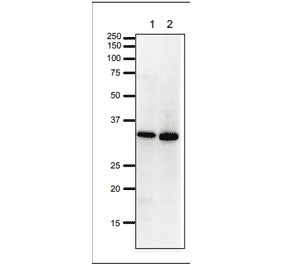
![Immunohistochemistry - Anti-PCNA Antibody [SPM350] - BSA and Azide free (A249579) - Antibodies.com](https://cdn.antibodies.com/image/catalog/252/A252759_1.jpg?profile=product_alternative)