

Unconjugated
Pathological cardiac hypertrophy often leads to heart failure. Activation of autophagy has been shown in pathological hypertrophic hearts. Autophagy is regulated positively by Class III phosphoinositide 3-kinase (PI3K). However, it is unknown whether Class III PI3K plays a role in the transition of cardiac hypertrophy to heart failure. To address this question, we employed a previously established cardiac hypertrophy model in heat shock protein 27 transgenic mice which shares common features with several types of human cardiomyopathy. Age-matched wild-type mice served as control. Firstly, a prolonged activation of autophagy, as reflected by autophagosome accumulation, increased LC3 conversion and decreased p62 protein levels, was detected in hypertrophic hearts from adaptive stage to maladaptive stage. Moreover, morphological abnormalities in myofilaments and mitochondria were presented in the areas accumulated with autophagosomes. Secondly, activation of Class III PI3K Vacuolar protein sorting 34 (Vps34), as demonstrated by upregulation of Vps34 expression, increased interaction of Vps34 with Beclin-1, and deceased Bcl-2 expression, was demonstrated in hypertrophic hearts from adaptive stage to maladaptive stage. Finally, administration with Wortmaninn, a widely used autophagy inhibitor by suppressing Class III PI3K activity, significantly decreased autophagy activity, improved morphologies of intracellular apartments, and most importantly, prevented progressive cardiac dysfunction in hypertrophic hearts. Collectively, we demonstrated that Class III PI3K plays a central role in the transition of cardiac hypertrophy to heart failure via a prolonged activation of autophagy in current study. Class III PI3K may serve as a potential target for the treatment and management of maladaptive cardiac hypertrophy.
Proteasomes are attractive emerging targets for anti-cancer therapies. Auranofin (Aur), a gold-containing compound clinically used to treat rheumatic arthritis, was recently approved by US Food and Drug Administration for Phase II clinical trial to treat cancer but its anti-cancer mechanism is poorly understood. Here we report that (i) Aur shows proteasome-inhibitory effect that is comparable to that of bortezomib/Velcade (Vel); (ii) different from bortezomib, Aur inhibits proteasome-associated deubiquitinases (DUBs) UCHL5 and USP14 rather than the 20S proteasome; (iii) inhibition of the proteasome-associated DUBs is required for Aur-induced cytotoxicity; and (iv) Aur selectively inhibits tumor growth in vivo and induces cytotoxicity in cancer cells from acute myeloid leukemia patients. This study provides important novel insight into understanding the proteasome-inhibiting property of metal-containing compounds. Although several DUB inhibitors were reported, this study uncovers the first drug already used in clinic that can inhibit proteasome-associated DUBs with promising anti-tumor effects.

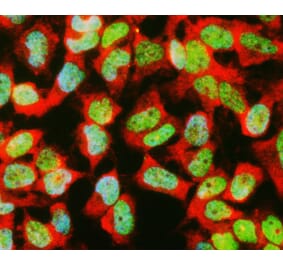
![Immunofluorescence - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_1.jpg?profile=product_alternative)
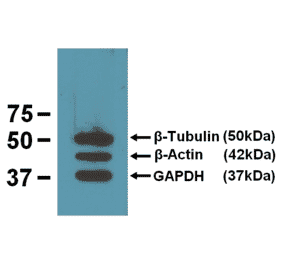
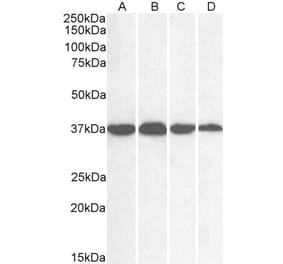
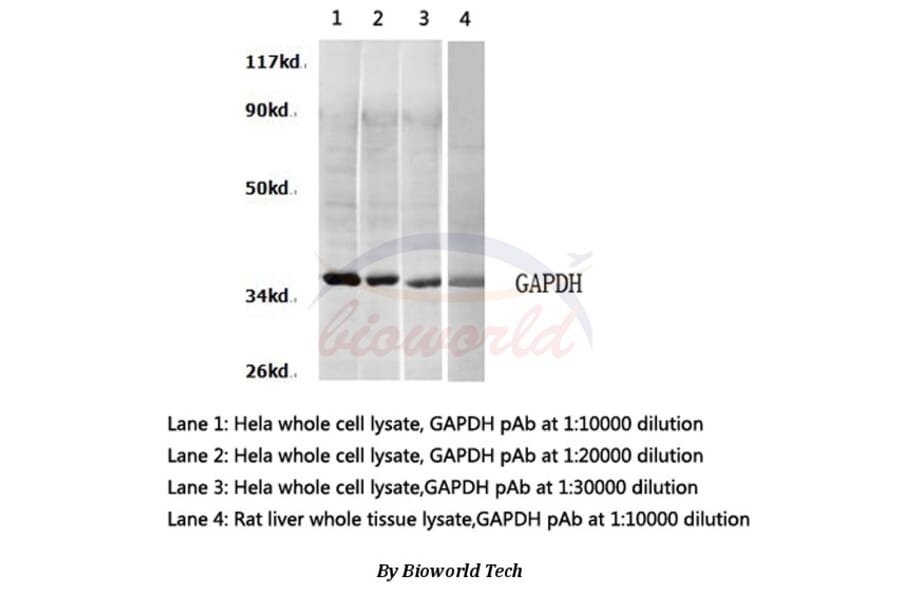















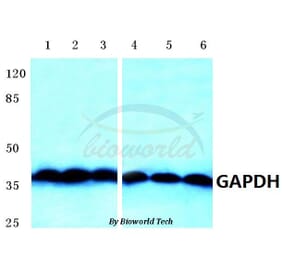
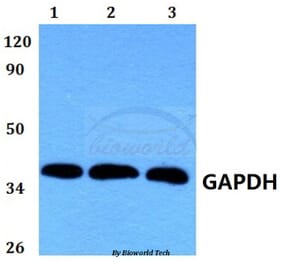
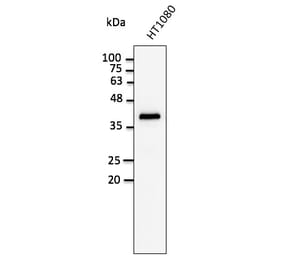
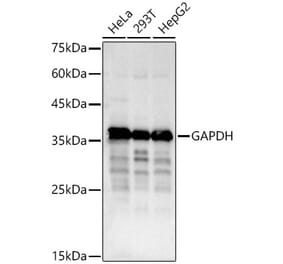

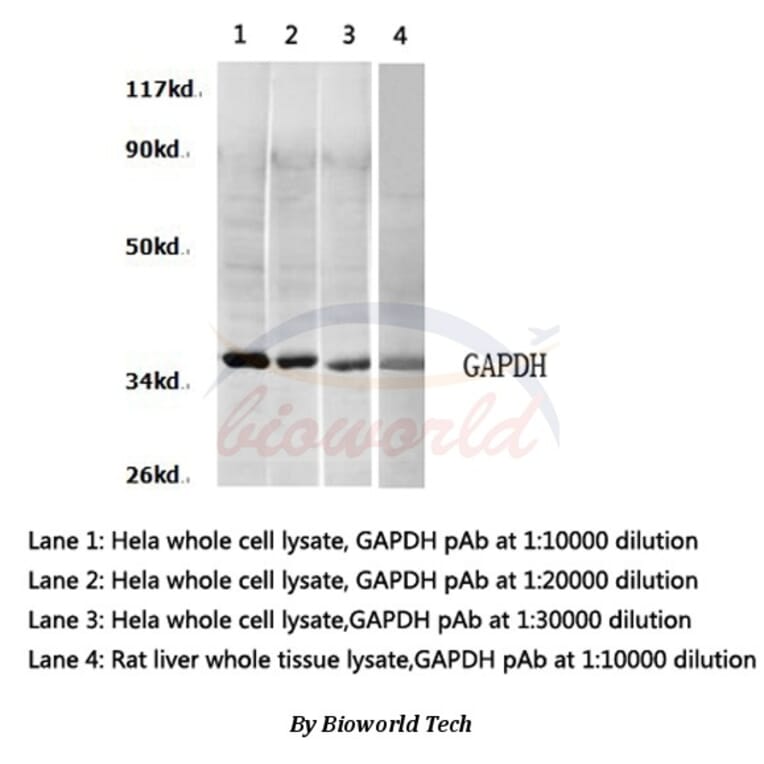
![Western Blot - Anti-GAPDH Antibody [ARC50888] (A309068) - Antibodies.com](https://cdn.antibodies.com/image/catalog/309/A309068_1.jpg?profile=product_alternative)
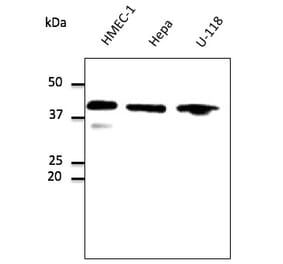
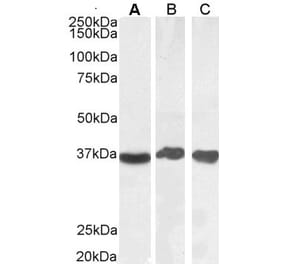
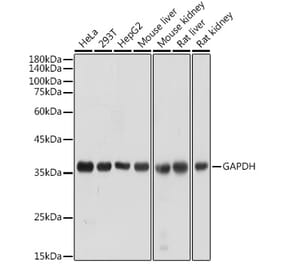
![Western Blot - Anti-GAPDH Antibody [AMC0062] (A92903) - Antibodies.com](https://cdn.antibodies.com/image/catalog/92/A92903_1.jpg?profile=product_alternative)
![Western Blot - Anti-GAPDH Antibody [AMC0062R] (A17301) - Antibodies.com](https://cdn.antibodies.com/image/catalog/17/A17301_1.jpg?profile=product_alternative)