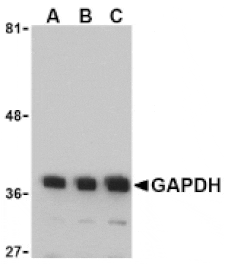
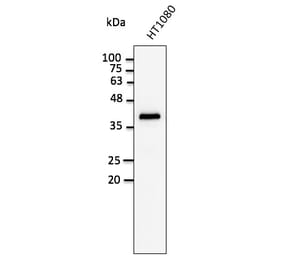
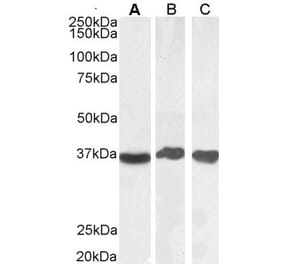
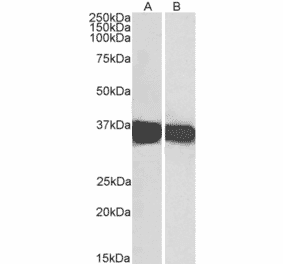
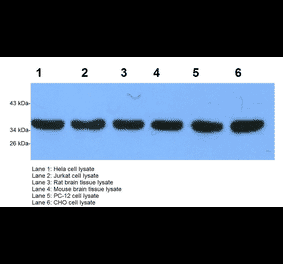
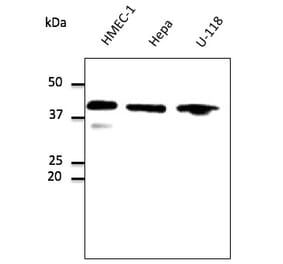
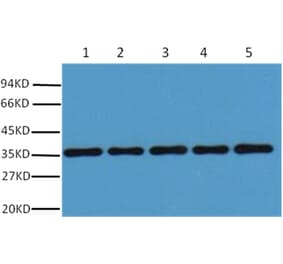
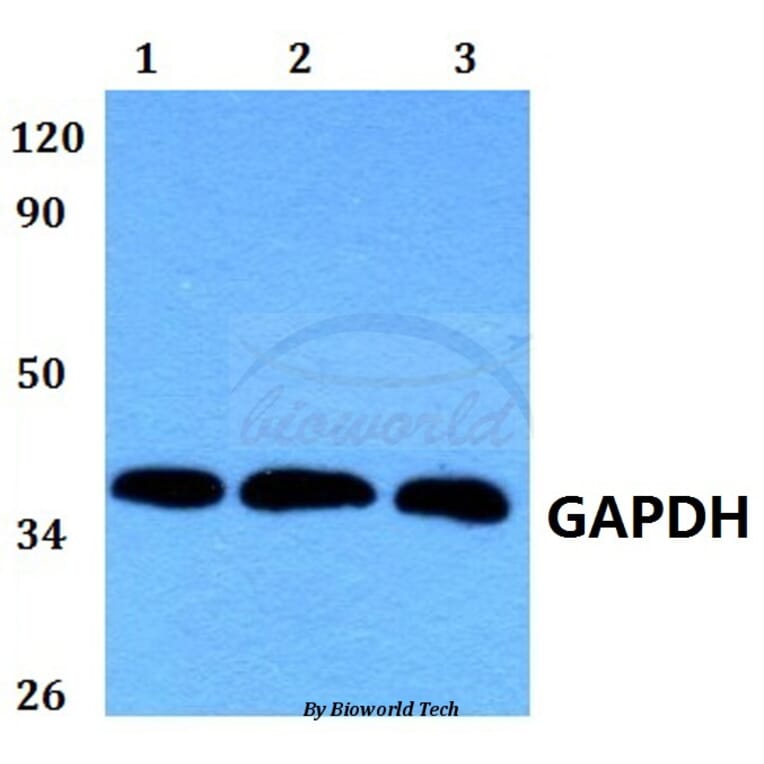
GAPDH (1A6) mAb detects endogenous levels of GAPDH protein.
Applications
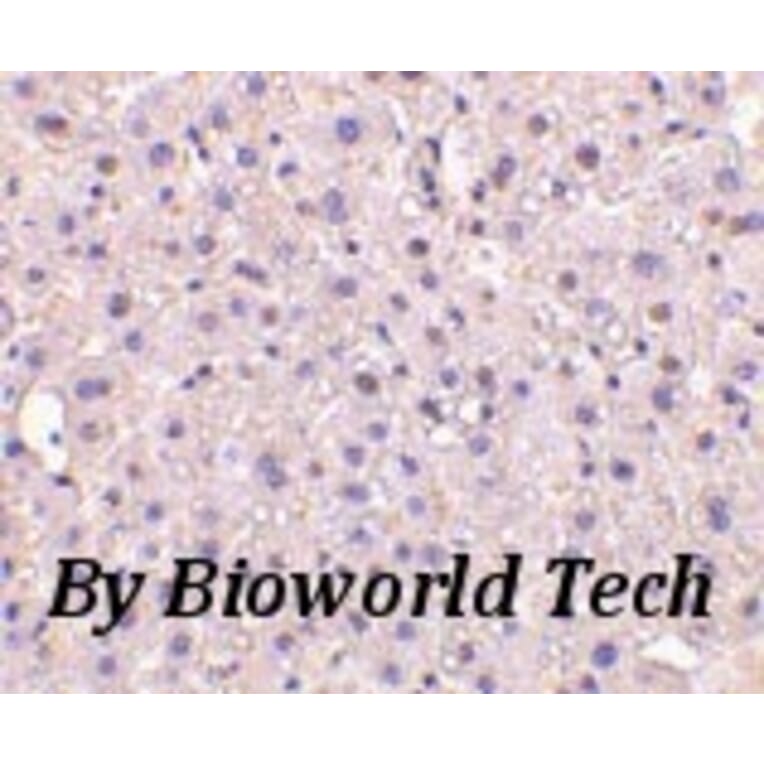
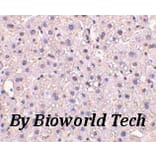
ELISA, WB, IHC, IF
Reactivity
Human, Mouse, Rabbit, Frog, Fish, Chicken, Rat
Immunogen
Recombinant full length Human GAPDH.
Host
Mouse
Clonality
Monoclonal
Conjugate
Unconjugated
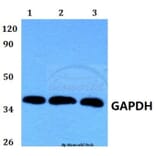
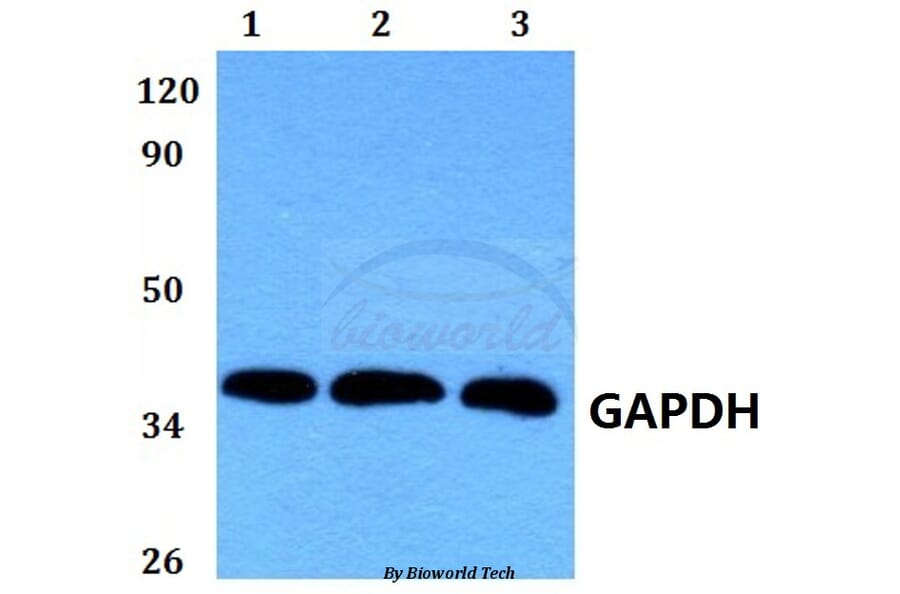
Molecular Weight
~ 36 kDa
Purity
The antibody was affinity-purified from mouse antiserum by affinity-chromatography using epitope-specific immunogen and the purity is > 95% (by SDS-PAGE).
Product Form
1 mg/ml in Phosphate buffered saline (PBS) with 15 mM sodium azide, approx. pH 7.2.



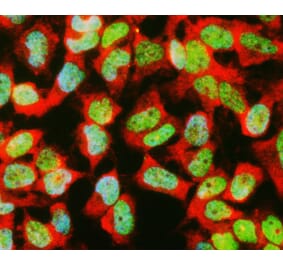
![Immunofluorescence - Anti-GAPDH Antibody [1D4] (A85382) - Antibodies.com](https://cdn.antibodies.com/image/catalog/85/A85382_1.jpg?profile=product_alternative)
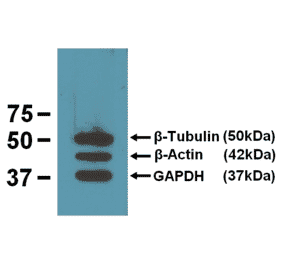

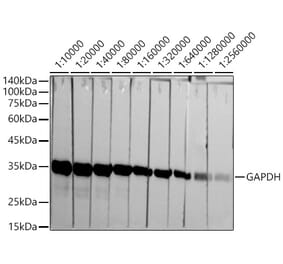
![Western Blot - Anti-GAPDH Antibody [6C5] (A279474) - Antibodies.com](https://cdn.antibodies.com/image/catalog/279/A279474_4.jpg?profile=product_alternative)
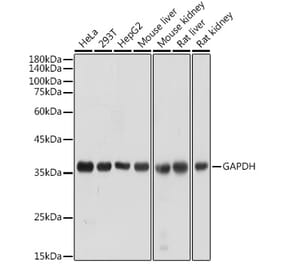
![Western Blot - Anti-GAPDH Antibody [4G5] (A279385) - Antibodies.com](https://cdn.antibodies.com/image/catalog/279/A279385_5.jpg?profile=product_alternative)